Project Description: Improving DNA Storage with Synthetic Biology
Overview
With the world entering the zettabyte era, the current data storage model that relies heavily on ‘the cloud,’ or large data storage centers, is projected to be incapable of meeting society’s growing demand in data storage. To give everyone some context, 1 zettabyte is equivalent to 1 trillion gigabytes.
Our project aims to tackle the growing need for a better, more energy-efficient data storage medium compared to current magnetic and optical data storage options by means of synthetic biology. Currently, we aim to achieve this through 2 separate tracks:
- Developing an enzymatic DNA synthesis platform that can elongate a single-stranded DNA (ssDNA) in a template-independent manner. The synthesized ssDNA strand will then be converted to a more stable, double-stranded DNA (dsDNA) and inserted into a plasmid for long-term data storage.
- Developing a data encoding/decoding pipeline that allows binary files (used by computers) to be stored in a ternary format compatible with our DNA synthesis platform, retrieved, and converted back into binary.
Context and Scope
How we decide to shape the context of our project will largely depend on the iHP interviews our team will be conducting throughout the season. Depending on our project’s ‘story,’ the technical aspects of our project’s design may also change to reflect the story’s emphasis. Below are 3 potential contexts in which our project can be placed as of now:
- Preserving indigenous stories
- Storing archived governmental records
- Storing archived medical records
The technical scope of our project is mostly limited by the iGEM competition timeline; we essentially have 4 months to carry out most of our wet lab experiments and produce a minimum viable product (MVP). This means that our project’s aim is to:
- Successfully demonstrate a proof-of-concept DNA synthesis and storage platform with sample data relevant to our project’s context.
- Successfully perform E-DBTL cycles from both wet lab and dry lab to convince the iGEM judges that our project’s design went through multiple iterations that each develop on top of each other.
Note that our project does NOT aim to:
- Develop a polished, complete DNA synthesis and storage platform that is ready for launch. We will not have the time nor resources to make this happen by the Jamboree. Keep this in mind when you’re working on the project and try not to focus on details that are not required for a proof-of-concept or an MVP.
Current Plans
DNA Synthesis Platform
We will be using terminal deoxynucleotidyl transferase (TdT) to enzymatically synthesize ssDNA strands since TdT does not require a template strand for DNA elongation; it just requires a short nucleotide sequence (a primer) to add nucleotides to at the 3’ OH position. When natural deoxynucleoside triphosphate (dNTP) - guanine (dGTP or G), cytosine (dCTP or C), adenine (dATP or A), and thymine (dTTP or T) - is provided to TdT, the enzyme will perform ssDNA elongation until it either runs out of dNTPs to add or if the reaction condition is no longer favorable. In other words, even if we just provide 1 type of dNTP for TdT to work with, we cannot control the number of nucleotides added to the strand in a single reaction cycle (reagent addition → incubation → reagent wash), making our synthesis method ‘semi’-specific.
TdT’s ssDNA elongation efficiency also decreases when the ssDNA strand starts to fold on itself and create secondary DNA structures (i.e., loops and folds). To prevent this, we can increase the reaction temperature as secondary DNA structures are much less likely to form at higher temperatures. Natural or wildtype (WT) TdT, however, cannot withstand such high temperatures. We aim to resolve this issue by using a modified thermostable TdT (TS TdT) that can withstand higher temperatures. By synthesizing ssDNA strands using TS TdT and higher reaction temperatures, we aim to increase the synthesis efficiency of our platform.
We will be performing ssDNA elongation with TdT on a solid phase synthesis (SPS) platform. Unlike Aachen 2021’s method where an immobilized ssDNA primer was ‘dipped’ into multiple reaction tubes, we will be immobilizing our ssDNA primer on a solid plate (either made of glass or plastic) and have our reagents flow to the plate’s surface.
Software
Although TdT is capable of adding all 4 types of dNTPs, it is known to prefer adding nucleotides to certain primer sequences that meet specific constraints. We will be developing a software algorithm that can generate the ‘best’ candidate primer sequences that can be used to initiate ssDNA elongation. We will also be developing a complete data encoding and decoding software pipeline that will convert a binary input file (compatible with computers) into a ternary representation of data to be stored in DNA sequences (to be synthesized with our platform) with corresponding metadata, then retrieve and decode the sequence information back into binary for users to access the stored data. Note that our sequence will be encoding information in ternary (using 0, 1, and 2) instead of quaternary (using 0, 1, 2, and 3 like the 4 types of dNTPs) due to our synthesis platform being semi-specific, as previously mentioned. Rather than having each dNTP correspond to a single number (such as G=0, C=1, A=2, T=3), we will be assigning a single number to the transition between 2 types of dNTPs (such as C→G=0, C→T=1, C→A=2). There will be various types of metadata assigned to each ternary DNA sequence, which will be used to identify where the retrieved information belongs to in our filing directory and help decode the sequence information back into binaries. We will also be implementing error correction algorithms in our decoding step to increase the accuracy of our retrieval process. We will also be developing a graphical user interface (GUI) to allow users to ‘upload’ and ‘download’ their files from our DNA-based data storage platform. Note that we will not be able to perform the complete workflow starting from file uploading, synthesis, retrieval, and downloading for all end-users at the Jamboree due to time and physical constraints. But the GUI will be a good visual representation of our end-goal and be integrated into our MVP for demonstration purposes.
Hardware
SPS is a synthesis method that is highly compatible with microfluidics, since they only differ in the reaction scale; SPS operates in a manual scale where the user directly drops or flows reagents to the immobilized ssDNA primer, while in a microfluidic chip the reagents flow through narrow channels in a much smaller scale (i.e., millimeters). Once we demonstrate that ssDNA can be elongated with TS TdT in an SPS platform, we will then be transferring that reaction into a microfluidic chip. This will allow our synthesis reactions to be parallel, automated, high throughput, and higher precision as microfluidic pumps used to control reagent flow through different channels can be controlled by a computer. We will also be building upon the bioreactor from the 2023 team to culture E. coli expressing TdT.
Human Practices
We want our project to be relevant for stakeholder use and have their perspectives incorporated into our project’s design through the entire engineering design, build, test, and learn cycle (E-DBTL cycle). We will be reaching out to various industry professionals and community members for advice and feedback on our project’s design throughout the season. We will also be planning multiple synthetic biology- and DNA data storage-related initiatives to raise our profile.
Main Goals and Side Goals
Below is a list of our main project goals that are required for our team to win a Gold medal at the Jamboree:
- Demonstrate that an SPS platform can be used to elongate ssDNA strands with TdT
- Express TS TdT using site-directed mutagenesis from WT TdT in E. coli
- Optimize the reaction conditions for TS TdT in an SPS platform
- Develop a primer generation algorithm to identify optimal ssDNA primer sequences
- Develop a pipeline to encode data into DNA sequences/corresponding metadata
- Develop a pipeline to decode DNA sequences using error correction algorithms and metadata
- Develop a graphic user interface (GUI) that demonstrates how users can upload and download their files from our system
- Design a microfluidic chip that translates TS TdT-based ssDNA elongation in an SPS platform into the microfluidic scale
- Build upon the current bioreactor to allow TdT culturing
- Demonstrate the E-DBTL cycle within our experimental, software, and hardware aspects of the project
- Incorporate feedback and advice from industry professionals and stakeholder communities into our project design and development
Other Implementations
Aachen 2021 team also developed an enzymatic DNA synthesis platform - the DIP method -, where immobilized primer strands are dipped into reaction tubes each containing TdT and 1 type of dNTP. Several points where our system differs from theirs is:
- We aim to develop an SPS platform, where the immobilized primer isn’t exposed to the stress of being physically moved around
- We will be using TS TdT instead of WT TdT for enzymatic DNA synthesis
- We will be incorporating more error correction measures to increase the accuracy of data recovery
There is also published literature 1, 2, 3 that looks into TdT-based enzymatic DNA synthesis, but most of their techniques have been patented.
Attributions
iGEM requires everyone who contributed to the project to fill out the attributions form. For members on the team, leads and co-directors will be checking the Master Tracker to verify that your attributions are honest. Wording is important; don't say more than what you actually did.
How to fill out attributions
Go here to figure out what to put in each column.
Internal Contributions
PI and Co-Directors
| Name | Role | General Tasks | Specific Tasks |
|---|---|---|---|
| Steven Hallam | Primary PI | ||
| Chaehyeon Lee | Student Leader | Conceptualization, Fundraising, Project Administration, Background Research | (1) Conceptualization - Facilitated the team-wide project brainstorming process. (2) Fundraising - Supervised and led various fundraising initiatives including Departmental, Faculty, and Industry sponsorship requests. (3) Project Administration - Supervised and led the project's overall progress and administrative tasks. (4) Background Research - Organized weekly project development meetings during March to ensure subteam integration during the project design phase. Performed literature review to ensure project's experimental plan is feasible within the competition timeline. Reached out to various external advisors for technical advice to ensure project is feasible. |
| Narjis Alhusseini | Student Leader | Conceptualization | (1) Conceptualization - Facilitated the team-wide project brainstorming process. |
Wet Lab
| Name | Role | General Tasks | Specific Tasks |
|---|---|---|---|
| Tina Wang | Student | Conceptualization, Background Research, Investigation, Notebook/recard keeping Project Administration | |
| Achint Lail | Student | Fundraising, Conceptualization | (1) Fundraising - Reached out to industry and academic sponsors to secure grants and funding. (2) Conceptualization - Brainstormed potential TdT enzymes to explore and attributes to insert into the plasmid |
| QingRu Kong | Student | Conceptualization, Background Research, Investigation, Safety, Project Administration, Notebook/record keeping | |
| Ada Jiang | Student | Conceptionalization | (1) Conceptionalization - Brainstorming and developing ideas for the project |
| Burak Ozkan | Student | Background Research, Conceptualization | (1) Background Research - helped contribute to literature review regarding our probiotic to reduce emissions from cows and the wildfire pitch to study A. vinelandii, (2) Conceptualization - helped come up with the probiotic idea and support the importance of the organism in the pitch |
| Chloe King | Student | Conceptualization | (1) Conceptualization - Brainstorming and developing ideas for the project |
| Daniel Hinatsu | Student | Conceptualization | (1) Conceptualization - Learned how to update the wiki, brainstromed the final project |
| Diego Perez Hidalgo | Student | Conceptualization | (1) Conceptualization - |
| Pattarin Blanchard | Student | Conceptualization, Background Research | (1) Conceptualization - Contributed to project pitch at later stages; (2) Background Research - Literature review for TdT variants, 3'-protected, fluorescent-tagged dNTPs suitable for de novo enzymatic DNA synthesis |
| Ran Tao | Student | Conceptualization, Background Research, Investigation |
Dry Lab
| Name | Role | General Tasks | Specific Tasks |
|---|---|---|---|
| Piyush Awasthi | Student | ||
| Lucy Hao | Student | Notebook/record keeping, Project Administration, Wiki Coding, Writing, Background Research, Conceptualization | (1) Notebook/record keeping - Standardized and maintained laboratory notebook through creation, implementation of internal wiki for team. Took weekly meeting notes for dry lab and wiki meetings. (2) Project Administration - As dry lab co-lead, organized subteam members into projects and monitored progress. Liaison between team and PI. (3) Wiki Coding - As wiki lead, is the primary developer of the competition wiki, leading and overseeing all development and content that is added to the wiki by other subteam members. (4) Writing - (5) Background Research - (6) Conceptualization - |
| Matthias Wong | Student | Conceptualization | (1) Conceptualization - |
| Riya Alluri | Student | Conceptualization | (1) Conceptualization - Contributed to brainstorming for initial project ideas and developing final project pitch. |
| Samuel Salitra | Student | Notebook/record keeping | |
| Sebastian Hyland | Student | Conceptualization | (1) Conceptualization - Brainstormed software encoding/decoding/GUI portions of DNA data storage pitch |
HP and Design
| Name | Role | General Tasks | Specific Tasks |
|---|---|---|---|
| Harrison Kim | Student | Conceptualization, Public Engagement | (1) Conceptualization - helped with initial brainstorming of our project; (2) Public Engagement - Helped development of our blog and start our educational initiative |
| Yejin Lhee | Student | Conceptualization | (1) Conceptualization - Ideated HP tasks for the main project. |
| Charlotte Lee | Student | Conceptualization | (1) Conceptualization - brainstorming and developing ideas for the project |
| Claire Pinckney | Student | Conceptualization | (1) Conceptualization - Ideated HP tasks for the main project. |
| Jessica Xin | Student | Conceptualization, Public Engagement | (1) Conceptualization - helped brainstorm ideas for project; (2) Public Engagement - created hands on synbio acitivites for high school students; |
| Aoniya Colynn | Student | Conceptualization, Visualization | (1) Conceptualization - Brainstorming; (2) Visualization - Social media graphics. |
| Karen Lin | Student | Visualization | (1) Visualization - created logo for simply synbio |
Internal Advisors
| Name | Role | General Tasks | Specific Tasks |
|---|---|---|---|
| Brian Guo | Conceptualization | Gave feedback during project pitch brainstorming. | |
| Janella Schwab | Other | Helped with getting us in contact with Eric Ma, one of the founding members of UBC iGEM | |
| Laura Gonzalez Campos | Conceptualization | Gave feedback during project pitch brainstorming. | |
| Madina Kagieva | Conceptualization | Gave feedback during project pitch brainstorming. | |
| Nikita Telkar | Conceptualization | Gave feedback during project pitch brainstorming. | |
| Rodrigo Vallejos | Conceptualization | Gave feedback during project pitch brainstorming. | |
| Anjali Parthasarathy | Safety | Trained wet lab leads and codirectors. | |
| Brenda Ma | Conceptualization | Gave feedback during project pitch brainstorming. | |
| Edward Li | Conceptualization | Gave feedback during project pitch brainstorming. | |
| Parneet Sekhon | |||
| Umar Ali |
External Contacts/iHP
As we start reaching out to external contacts who are willing to help us, once they have given us advice, please either
- make an issue to track their name down in our internal wiki so Lucy or a wiki liaison can add them to the attributions page; you can make another issue to add updates to the external contact throughout the season
- or directly edit the iHP page yourself with your external contact and update that entry throughout the season.
The earlier we record down an external contact the easier it is to track so we can add them to our competition wiki.
Wet Lab
Notebook
For each notebook entry, copy the template here. Additionally, add a weekly summary to the main notebook page.
Goals
- Work as a team through the DBTL cycle
- Practice experimental planning, analysis and data collection
- Learn how to interpret this data for our Proof of Concept and to tell a story on our Wiki
- Learn essential lab skills for synthetic biology
- Adhere to lab safety and diligent note taking during experiments
- Develop problem-solving skills to identify and address issues that may arise during experiments
- Foster collaboration and communication with other sub teams
Timeline
| Event | Start Date | End Date |
|---|---|---|
| Project pitch brainstorming | January | |
| Lab inventory (Leads) | January | |
| Wet lab workshop on relevent knowledge and techniques | February | |
| Project brainstorming + propose wet lab projects | February | March |
| Assign individual tasks | April | |
| In-lab experiments | May | August |
iGEM Medal Requirements
Bronze
-
Project description:
- Describe how and why you chose your iGEM project
-
Contribution:
-
Make a useful contribution for future iGEM teams.
-
Add new documentation to an existing Part on that Part's Registry page:
- New information learned from literature
- New data collected from laboratory experiments
- Document troubleshooting that would be helpful to future teams
-
Add new documentation to an existing Part on that Part's Registry page:
-
Make a useful contribution for future iGEM teams.
Silver
-
Engineering success:
-
Go through at least one iteration of the engineering design cycle:
- Design → Build → Test → Learn
-
Go through at least one iteration of the engineering design cycle:
Gold
-
Excellence in Synthetic Biology
- General Biological Engineering
- and in at least one Specialization
Notebook: Weekly Summaries
Please add a weekly summary to this page.
April
Design
Why these projects?
Projects Overview
TdT Production
| Component | Priority | Objective | Assigned to | Report to |
|---|---|---|---|---|
Solid Phase Synthesis
| Component | Priority | Objective | Assigned to | Report to |
|---|---|---|---|---|
ssDNA to dsDNA
| Component | Priority | Objective | Assigned to | Report to |
|---|---|---|---|---|
Solid phase DNA Synthesis
- Context and Scope
- Goals
- The actual design of immobilization
- The actual design of modified DNTPs
- Proposed solutions to immobilization
This document outlines the process of non-traditional solid phase DNA synthesis through the use of tdt. In particular, this document will provide a broad overview of the workflow and the details of primer immobilization, solid phase chemistry, and primer cleavage. It also outlines the chemistry of monomers intended to use.
Context and Scope
Traditional solid phase chemical synthesis occurs from 3’ -> 5’ as shown below.
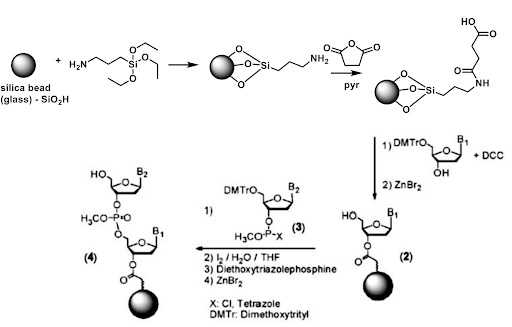
This is an important consideration as a robust set of chemical reactions exist for the immobilization of nucleic acids on the solid phase. However, there is a comparatively smaller tool box for synthesis in the reverse (5’->3’) direction. Additionally, chemical solid phase synthesis involves protecting groups on the exocyclic nitrogenous bases. For our purposes, we require a method to immobilize the 5’ end on the solid phase, leaving the 3’ end open to react with incoming DNTPs.
For controlled single nucleotide insertion, a robust yet readily cleavable protecting group is required. The primary method of 3’ protection for 5’ to 3’ synthesis is photolabile protecting groups. These groups are efficiently cleaved when exposed to UV light. These groups have been developed due to the desire to generate complex microarrays for genetic analysis. With photolabile protecting groups, a desired pattern of unique oligonucleotides may be synthesized. This chemistry can be leveraged for the use of TDT single nucleotide insertion.
Goals
The goal of this phase is
- To functionalize the surface for the immobilization of primers
- To immobilize the primer 5’ end on the solid phase at a high enough density and selectivity to enable efficient DNA synthesis
- To characterize the density of primer immobilization
- Lastly, the primer and newly synthesized DNA strand must be cleaved from the solid phase
The actual design of immobilization
There are two different methods being considered for immobilization. All approaches are based on the same principle.
Disulfide 5’ modifier and dibenzocylooctyne 5’ modifier are the primer methods in consideration. The idea with each modified primer is to use orthogonal reacting functional groups. As a DNA oligo has both a 3’ and 5’ free hydroxyl group, using a 5’ modifier that selectively reacts with its counterpart would prevent the 3’ hydroxyl from reacting in any meaningful quantities.
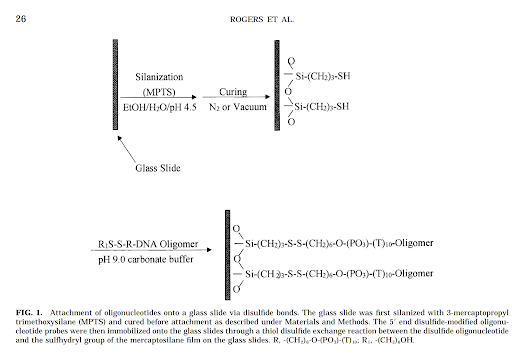
Rogers, Y.-H.; Jiang-Baucom, P.; Huang, Z.-J.; Bogdanov, V.; Anderson, S.; Boyce-Jacino, M. T. Immobilization of Oligonucleotides onto a Glass Support via Disulfide Bonds: A Method for Preparation of DNA Microarrays. Analytical Biochemistry 1999, 266 (1), 23–30. DOI:10.1006/abio.1998.2857.
The actual design of modified DNTPs
The DNTPs will be ordered as 3’-BzNPPOC modified DNTP’s or 3’aminohydroxy modified DNTPs. BzNPPOC is a highly photolabile group that is readily cleaved in UV light. This protecting group has been highly used in chip chemistry however care is required to prevent premature deprotection when exposed to ambient light. 3’aminohydroxyl groups are readily cleaved in sodium nitrite. These modified nucleotides will allow for single insertion as there is no 3’ hydroxyl for additional reaction. Thus a specific synthesis can be achieved. As part of the tdt synthesis workflow, an additional deprotection step is required prior to the following nucleotide reaction.
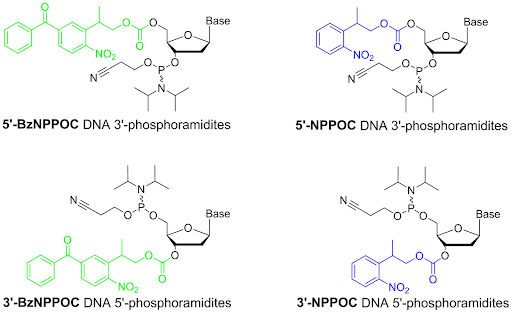
Hölz, K.; Hoi, J. K.; Schaudy, E.; Somoza, V.; Lietard, J.; Somoza, M. M. High-Efficiency Reverse (5′→3′) Synthesis of Complex DNA Microarrays. Scientific Reports 2018, 8 (1). DOI:10.1038/s41598-018-33311-3.
Verardo, D.; Adelizzi, B.; Rodriguez-Pinzon, D. A.; Moghaddam, N.; Thomée, E.; Loman, T.; Godron, X.; Horgan, A. Multiplex Enzymatic Synthesis of DNA with Single-Base Resolution. Science Advances 2023, 9 (27). DOI:10.1126/sciadv.adi0263.
Proposed solutions to immobilization
Initially, the glass will be functionalized or derivatized. The free hydroxyl groups react with a molecule containing a silyl ether on one end and a functionalized group on the other end.
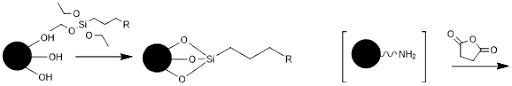
In the case of the 5’ disulfide modified primer, R would be a thiol. For the 5’ dibenzocylcooctyne modified primer, R would be an azide. This functionalizes the glass surface for addition of various chemicals. Addition of the 5’ modified primer would then react with the glass surface and covalently immobilize it.
The immobilization will be tested with fluorescing primers. Primers used for optimization will require both the necessary 5’ modifier as well as a fluorescing group either on the 3’ end or nitrogenous base. Regardless of where the group is located, this will allow us to determine the efficiency of coupling as well as the density of primers.
The stability of the immobilization can also be tested against the flow of various reagents using this method.
Once the reagents are in position, the reactions and characterization should be complete within 2-3 weeks.
ssDNA to dsDNA Experimental Design
- Overview
- Context and Scope
- Goals
- Proposed solutions and workflow
- Plasmid integration
- How do we test this?
- How long will this take?
Overview
Convert ssDNA synthesized by thermostable TdT (mutant 3-2) to dsDNA to generate a more stable dsDNA to allow better long-term storage.
Context and Scope
Terminal deoxynucleotidyl transferase (TdT) is a special type of polymerase found in mammals that is able to synthesize ssDNA in a template-independent manner. Our project aims to utilize TdT to customize and synthesize DNA strands for data storage.
Since TdT’s ability is limited to ssDNA synthesis, while ssDNA is not a stable biological molecule, this constrains our ability to store data long term using ssDNA. Hence, we propose to convert ssDNA to dsDNA once it is synthesized by TdT.
Goals
- Synthesize dsDNA from ssDNA to achieve a more stable biological molecule for storage
Proposed solutions and workflow
PCR
-
Denaturation (~96°C): Melt secondary structure, linearize ssDNA
-
Annealing (~50-56°C): Binding of primer to ssDNA template
-
Extension (~72°C): Taq polymerase extends the primers → dsDNA
Primer design
Primer is designed to be complementary to the initiator DNA of ssDNA synthesis and polyA tail
Primer design requirement
- 40-60 GC%
- The forward and reverse primer can’t have a temperature difference of greater than 5C
- 15-25 nucleotides long
- Usually the melting temperature of the primer is 50-60C
- Avoid hairpin structure
Plasmid integration
Use cloning technique to integrate dsDNA into a plasmid in E.coli (PCR amplification, golden gate/Gibson assembly)
Depending on the vector we use:
- If we use type II restriction enzyme:
PCR amplify the restriction enzyme recognition site onto the donor sequence (dsDNA) then perform Golden Gate to digest and ligate the dsDNA into the plasmid in E.coli in a one-pot reaction
- If we use normal restriction enzyme:
-
PCR amplify complementary region of the plasmid sequence onto the donor sequence
-
Digest the plasmid with restriction enzyme
-
Perform Gibson Assembly to ligate the dsDNA into the plasmid in E.coli
Transformation
Integrate the plasmid into E.coli BL21 (DE3) from NEB for protein expression
Colony Picking
Pick colony with correct antibiotic resistance
How do we test this?
Options to see if this worked:
- Use the same PCR primer to amplify the dsDNA inside the plasmid then use agarose gel to measure the length of the sequence integrated
- Sanger sequence or NGS (depending on the situation)
How long will this take?
If everything goes wellwhile, 1-2 day. If not, 1 week should be enough for troubleshooting.
Thermostable TdT
- Overview
- Context and Scope
- Goals
- Design
- Protein Purification: Immobilized metal affinity chromatography
- How long will this take?
Overview
The document provides the details related to Terminal deoxynucleotidyl transferase (TdT) utilized in the project. This includes the general description of WT TdT and thermostable TdT, in addition to their application in the project.
Context and Scope
TdT is a specialized DNA polymerase that catalyzes the addition of nucleotides to the 3' terminus of a DNA molecule in a template-independent manner. This means it is able to synthesize single-strand DNA without an existing DNA strand as a template. This highlights its potential to be utilized as a biological tool to manipulate DNA synthesis, producing DNA strand as designed.
ssDNA is prone to secondary structure formation (1). This issue can be minimized when working under a higher temperature (1). Since wildtype TdT derived from mammals cannot function optimally under a higher temperature (>37°C), this reaction can be achieved using a thermostable TdT, which has a higher optimal activity temperature, which in our case, would be around 47°C.
Goals
- Clone thermostable TdT
- Produce and purify thermostable TdT
- Optimize TdT reaction condition
Design
Thermostable TdT Cloning: Ligation Independent Cloning (LIC)
Material:
- Plasmid of Choice: Addgene # 29659 - pET His6 sumo TEV LIC cloning vector
- Mutant TdT (purchased from IDT)
- BL21 (DE3) (puchased from NEB)
Procedure:
-
Primer Design
- Design forward and reverse primer for TdT on geneious
-
PCR Amplification
- Amplify the LIC fusion tag onto TdT sequence
-
Agarose Gel
- Gel purification and check if the correct base pairs sequence has been added onto TdT
-
Linearize the plasmid
- Use SspI restriction enzyme to linearize the vector
-
Gel Purification
- Gel purify the linearized product and if needed the PCR product
-
Ligation Independent Cloning
- Use T4 DNA Polymerase reaction with dCTP for insert and dGTP for vector
-
Transformation
- Transform chemically competent Top10 E.coli with cloning product
-
Colony picking
- Pick Kanamycin-resistant colonies
-
Miniprep
- Use Miniprep to isolate plasmid from E.coli
-
Sequencing
- Send plasmid for Sanger sequencing
Protein Purification: Immobilized metal affinity chromatography
Protocol reference link
TdT Optimization
Liquid Phase (Gel)
- Each reaction was carried out in 20µL total volume.
- All reaction components but the dNTP were assembled in 18µL dNTP was prepared in 2µL of water.
- The 18µL mix was composed such that upon mixing with the 2µL dNTP solution, the following initial composition would be obtained: 200µM dNTP, 1X TdT buffer, 0.05µM primer (TBD) 250µM cobalt chloride (if present), 1U/µL TdT
- To initiate the reaction, the 18µL mixture was added to a tube containing the 2µL dNTP mix and mixed immediately by pipetting.
- The reaction was then incubated at room temperature for at least two minutes, resolved on a TBE Polyacrylamide gel
- Length of ssDNA is determined by comparing with the ladder
Condition to compare: [@TdT_background]
Run reaction with each dNTP + ladder + primer reference
- Different dNTP concentration: 10, 25, 50, 100, 200, 400µM
- Different TdT concentration
- Different buffer concentration
- Different temperature: RT vs 37 vs 47 (mutant)
- With/without CoCl2: 0 vs 2.5mM vs 5 mM
- Different reaction time: 2 vs 10 vs 30 min
Testing dNTP concentration need for all 4 types of nucleotides The rest rxns can be carried with selected dNTPs
Protocol reference link
Solid Phase (TBD)
How long will this take?
This depends heavily on how successfully each experiment goes. The estimation is around 1 month.
Protocols
For each protocol, create a new file in SUMMARY.md. Include pictures, graphics, external sites to help explain your protocols to the other subteam members. Lots of protocols already exist on Google Drive. If they do, then link that protocol to the internal wiki. If you are designing a new protocol or editing an already existing protocol, please add it to the internal wiki. Lucy and wiki liaisons will be checking!
Engineering Success
Iteration 1
TdT Production
| Relevant Pages/Commits | Date(s) | |
|---|---|---|
| Design | March | |
| Build | ||
| Test | ||
| Learn |
Solid Phase Synthesis
| Relevant Pages/Commits | Date(s) | |
|---|---|---|
| Design | March | |
| Build | ||
| Test | ||
| Learn |
Parts
As you create and use parts, add them here.
Basic
| Part name | Type | Description | Length | Link on iGEM Registry |
|---|---|---|---|---|
Composite
| Part name | Type | Description | Length | Link on iGEM Registry |
|---|---|---|---|---|
Results
Safety
Human Practices
Goals
- Conduct iHP interviews to narrow our project
- Conduct education initiatives for a variety of age groups
- Primary school (CODE Initiative)
- Highschool (Let’s Talk Science, Geneskool Summer Initiative)
- Adult (Science 101)
- Continue our “Sustainable Development in Synthetic Biology” podcast
- Start our “Redefining Research” seminar series
- Continue our “Simply Synbio” blog with bi-weekly articles
- Create a user manual in parallel to our software project
Timeline
| Event | Start Date | End Date |
|---|---|---|
| Project pitch brainstorming | January | February |
| iHP Interviews | March | |
| Brainstorm Education, Inclusive, and Sustainability Initiatives | March | April |
| Let's Talk Science Classroom Visits | April | June |
| Reach out to Summer Education Initaitives | March/April | |
| Redefining Research Seminar Series | April | September |
| Inclusivity Initiatives | Summer | |
| Summer Camp | June | August |
| Sustainable Development in Synthetic Biology Podcast | April | August |
| Jamboree Preparation + Wiki Writing | September | October |
iGEM Medal Requirements
Bronze
- Project Description
-
Contributions Page
- Committing work to GitHub
- Documenting work relating to Human Practices
Silver
-
Explain how our work is responsible and good for the world
- What values did we have in mind when designing our project?
- Which resources or communities did we consult to ensure we have the appropriate values?
- What evidence is there that we thought about these things?
Gold
- Wiki Writing
-
Special Award Nomination
- Education
- Inclusivity
- Integrated Human Practices
Integrated Human Practices
This page contains all the external contacts who have helped shaped our project. For every entry, copy this template and fill out the TODOs.
Name
- person of contact: TODO
- institution type: TODO
- iHP interview notes: TODO
- relationship to team: TODO
- tasks: TODO
[TODO: few sentences on what they have helped us with]
Software
Dr. Jon Corbett
- person of contact: Narjis
- institution type: Academic
- iHP interview notes: 04/03/2024
- relationship to team: Collaborator, External Advisor
- tasks: Conceptualization
Provided context for our DNA storage platform (indigenous history) and offered technical advice for dealing with high rates of deletion
rmehri01
- person of contact: Lucy
- institution type: Industry
- relationship to team: External Advisor
- tasks: Wiki Coding, Conceptualization
Helped set up Codespaces for team to develop without needing to set up a local environment, as well as set of Replit. Ideation of algorithms for software and development stack.
Microfluidics
Dr. Albert Folch
- person of contact: Piyush
- institution type: Academic
- relationship to team: External Advisor
- tasks: Conceptualization
Helped dry lab with initial planning related to do SPS with microfluidics.
Dr. Karen Cheung, Professor at UBC SBME
- person of contact: Piyush
- institution type: Academic
- iHP interview notes: 28/03/2024
- relationship to team: External Advisor
- tasks: Conceptualization
Helping develop and refine microfluidic chips, primarily will be focusing on PCR chip.
Wet Lab
Name
- person of contact: TODO
- institution type: TODO
- iHP interview notes: TODO
- relationship to team: TODO
- tasks: TODO
- specific tasks:
- TODO
Ethics
Dr. Holly Longstaff
- person of contact: Yejin
- institution type: Industry
- iHP interview notes: 07/03/2024
- relationship to team: External Advisor
- tasks: Conceptualization
Helped give insight on the direction of the project related to health care data systems
Kenny Hammond
- person of contact: Jessica
- institution type: Industry
- iHP interview notes: 11/03/2024
- relationship to team: External Advisor
- tasks: Conceptualization
Helped give insight on the direction of the project related to health care data systems
Project Development
Dr. Nozomu Yachie, Professor at UBC SBME, Director of Research
- person of contact: Chae
- institution type: Academic
- iHP interview notes: 22/03/2024
- relationship to team: External Advisor
- tasks: Conceptualization
Provided technical advice during project conceptualization regarding the rationale behind using TdT-based enzymatic DNA synthesis, suggested using NGS to verify ssDNA elongation and strategic experimental design to overcome the long turnaround time between NGS sequencing runs, and suggested various encoding/decoding algorithms.
Eric Ma
- person of contact: Chae/Narjis
- iHP interview notes: 19/01/2024
- institution type: Industry
- relationship to team: Sponsor
Helping us pay for hosting of our domain, and gave general advice for managing a successful iGEM team
Communication
Infographics
Overview
From April to July, Design will make infographics regarding the topic to practice graphic creation in the context of science communication.
Context and Scope
Science communication and education are important in terms of creating an aware society that contributes to the science discourse. Infographics is one vessel that brings together aesthetic visuals and words to popularize knowledge regarding a certain topic.
The Design Subteam will create a series of infographics that reflect aspects of our project including, but not limited to: DNA Storage, the importance of science communication, microfluidics, and solid phase synthesis.
Goals
Goals:
- To create visually appealing, comprehensive infographics for the general public
- To practice making science-based graphics to convey information
Plan
- Design will write up the information on the infographics
- Any written information will go through approval from any related leads
- Planning sessions will be implemented to organize graphic creations as well as an understandable layout
- Final designs will be approved and shared to the rest of the team
- Infographics can be posted on our website + instagram (if possible)!
Blog
Overview
The Simply Synbio initiative from last year is introduced to a new medium of writing. It provides a way to bridge the gap between synthetic biology findings to the general public with the aims to promote discussion and invoke thought into the implications that synthetic biology discoveries brings to our communities.
Context and Scope
Science knowledge holds an important role in policy and decision making in society, which makes science communication crucial. The Simply Synbio blog is a team-managed blog that writes about new synthetic biology discoveries in a comprehensible format. The science blog is intended for audience members of all backgrounds to be able to understand and take part in the discussion of new breakthroughs with the science community. Along with the written portion of the blog, there is an even further condensed accompaniment posted on Instagram.
Goals and non-goals
Goals
- To communicate science to the general public
- To provide information in an accessible format; in a written form for the general public in mind and through further condensed Instagram posts
- Practice science communication among members
- Encourage members to partake in a team-wide activity
Non-Goals
- Increasing viewership and engagement - do not need to work on marketing other than reposting and supporting each other
Plan
All information is written in this document!
User Manual
Overview
The User Manual is a document that allows for scientists to use our product (DNA synthesis and software system) in an understandable and safe way.
Note that this is a working guideline; as the nature of the User Manual is based on an integration of all subteams, plans may change depending on the progress of Wet and Dry lab.
Context and Scope
As a foundational advance project with a clear deliverable in the form of a software that can be used to decode a DNA storage system, the deliverable should be as easy to handle as possible; in order to increase the accessibility and usability of our product, we need a manual that clearly describes the product in a step by step manner. The user manual should also include an extensive safety section in order to outline our deliverable as a safe product to use.
Goals and non-goals
Goals
- To clearly outline how to use our product
- To clearly outline the safety implications and hazards of the product
Non-Goals
- A summary of our whole project; engineering design process, wet lab details, human practices initiatives, should not be included
- Only relevant information will be included (context)
Plan
Sections to include in the document:
- Introduction
- Quick intro of our team + project
- Explain any important components (TdT, vectors, software)
- In what context the kit should be used
- Materials
- User Guide
- Biosafety
- Safety data sheet
- Handling information
Education
Internal Workshops
January
February
Git, GitHub, Software Component Workshop
- held by: Lucy
- slides
This workshop was held to teach everyone how to use Git and GitHub, so that everyone is able to contribute to the wiki that iGEM will be judging.
Dry Lab Integration
- held by: Lucy, Piyush
- slides
This workshop was held to introduce everyone to engineering cycles, and what dry lab plans to do this year.
March
Information Theory
- held by: Lucy
- slides
This workshop was held to give everyone some background information on the state of computers, DNA storage and a brief introduction to information theory.
April
May
Let's Talk Science
Overview
The collaboration between Let’s Talk Science and the UBC iGEM team is an educational outreach initiative designed to engage high school students in the exciting field of synthetic biology. Through interactive workshops, seminars, and hands-on activities, this initiative aims to foster greater interest in the field of synthetic biology and inspire the next generation of scientists.
Context and Scope
Let’s Talk Science’s extensive experience in science education combined with our UBC iGEM team’s research and practical knowledge in synthetic biology is ideal for sparking interest in the topic of synthetic biology for high school students. This program will focus on making advanced scientific concepts accessible and engaging to young learners across various educational backgrounds.
Goals and non-goals
Goals
- To increase awareness and understanding of synthetic biology among highschool students
- To inspire students to pursue further education and careers in the STEM fields, particularly in synthetic biology and related disciplines
Non-Goals
- To target audiences outside of the highschool education level for this specific initiative
- To provide university level technical training
Plan
Brainstorming and ideation of classroom activities
- Pitching idea to LTS representatives
- Marketing and branding
- Contacting highschools through Let’s Talk Science
- Volunteer training for our internal team
- Final activity preparations + material gathering
- Executing our program with classroom visits
OTHER PLANS NOT IN THE TIMELINE
Produce a DNA Storage specific classroom activity that complements our project
Summer Symposium
Overview
UBC iGEM team’s summer symposium initiative will invite high school students to pitch their innovative synthetic biology ideas, fostering a platform for creativity, learning, and exchange. This event will aim to connect young minds with experts in the field, encouraging discussion, feedback, and the development of students’ concepts in synthetic biology.
Context and Scope
Centered around a symposium format, this initiative allows students to showcase their ideas in front of a panel of synthetic biology experts and peers, promoting an environment of learning and constructive critique. This event will be tailored specifically for high school students interested in synthetic biology, aiming to bridge the gap between academic knowledge and real-world application.
Goals and non-goals
Goals
- To stimulate interest in synthetic biology among high school students
- Allow the creative process of idea generation and pitching
- Provide a supportive platform for students to receive expert feedback
- Enhance learning experience
- Encourage further exploration of STEM fields
Non-Goals
- Providing in-depth research opportunities
- Direct implementation of the pitched ideas
- Extending the initiative to include participants beyond high school students
- To cover areas outside of synthetic biology
Plan
- Invite high school students during Let’s Talk Science visits
- Host potential workshops focused on the fundamentals of synthetic biology and effective pitch techniques (can be done via Zoom)
- Have students submit their synbio project ideas
- Review submissions and give feedback
- Finalize logistics for the symposium
- Host the Summer symposium!
Inclusivity
Event Recaps
EDI Seminar: Redefining Research
EDI Seminar
Overview
Science is a collaborative process; this means that inclusivity to provide as many perspectives as possible is needed. Human Practices will focus on the aspects of what it means to uphold Human Practice values in a research setting by hosting a panel discussion with members of the EDI community.
Context and Scope
Decolonizing research and being inclusive is needed in the science community in order to create a platform where ideas are shared equitably and fairly; this ensures that all voices are heard and contributes to discussion regarding societal and ethical implications of science.
Human practices will be leading an EDI Panel discussion that focuses on Human Practices values.
Goals and non-goals
Goals
- To promote a platform of discourse on what it means to practice humanity within research
- To promote discussion on how we can focus research on inclusivity; both within the research community as well as the wider community
- To highlight voices that may have been previously marginalized
- To discuss ways that we can make research more accessible and inclusive; decolonizing research
- To provide a safe space where natural and authentic discussion can happen
- Encourage the thinking of the implications of science
Non-Goals
- To enforce ideas on others - this platform lends itself for us to bring the idea of Human Practices onto the table; it is up to the panel to do with the information as they wish
Plan
- We will invite 2-4 guest speakers with EDI backgrounds to come in to the seminar
- The first part of the seminar will be consist of an introduction for everyone
- The professors and their research
- The Human Practices team and what Human Practices is
- The second part will lend itself as a platform for discussion on what Human Practices is, guided by questions created by the Human Practices subteam
- After the premeditated questions are over, questions can be asked by the audience
- After the session is over and time and venue allows, mingling may be allowed.
Sustainability
Sustainable Development in Synthetic Biology
Overview
The “Sustainability in Synthetic Biology” podcast is an engaging platform where academic, industry, and community professionals are invited to discuss the United Nations Sustainable Development Goals (SDGs) through the lens of synthetic biology. Each episode will focus on 1-3 SDGs, exploring how innovations in synthetic biology can contribute to achieving these goals.
Context and Scope
This initiative will build on the success of the previous two years that it has been run, aiming to deepen the conversation around the SDGs by connecting listeners with experts from diverse fields. The podcast will not only educate its audience on the potential of synthetic biology in addressing global challenges but also foster a community of like-minded individuals' passion about sustainable development.
Goals and non-goals
Goals
- To continue exploring the intersection of synthetic biology with the SDGs
- Enrich the audience’s understanding and appreciation for the field’s potential to impact global sustainability positively
- To cultivate meaning connections
- To gain valuable relationships with influential health professionals
- To establish a network of recurring and new guests who can contribute valuable insights to the podcast
Non-Goals
- To venture into discussions that do not have a clear link to these areas
- Increasing viewership and using it for funding purposes
Plan
- Identify and prioritize SDGs that have not been covered in previous sessions or new advancements
- Brainstorm SDGs that can also be project specific
- Reach out to previous guests who have been on the podcast before as well as new voices and experts who can bring unique perspectives to the podcast.
- Figure out how to enhance production quality (better question, recording, editing, design, etc)
- Implement a feedback mechanism for listeners and guests (identify opportunities for growth and improvement in future episodes)
Dry Lab
Goals
-
Create a layman's version of the project from a synthetic biology perspective for dry lab members.
-
Design all dry lab projects to have a clear and meaningful integration to another sub team.
-
Have a constant feedback loop with wet lab team.
-
Engage in outreach events with human practices.
Timeline
| Event | Start Date | End Date |
|---|---|---|
| Project pitch brainstorming (✔️) | January | February |
| Assessment of all dry lab team members to projects (✔️) | February | March |
| Start thinking of potential dry lab projects (✔️) | February | April |
| Search for resources, advisors, etc (Leads initially, later whole team) | March | |
| Assign self learning tasks (Leads) | March | |
| Finalize dry lab projects (Make sure connection to wet lab is clear) | March | April |
| Individual projects begin | April | August |
| Project check-in #1 | April | |
| Wiki Writing (GitLab Repo is provisioned) | June | September |
| Project check-in #2 | June | |
| Project check-in #3 | July | |
| Project check-in #4 | August | |
| Jamboree Preparation | September | October |
iGEM Medal Requirements
Bronze
-
Contributions Page
- Committing work to GitHub
- Documenting work relating to hardware
Silver
-
Engineering Success
-
Go through at least one iteration of the engineering design cycle:
- Hardware: Design → Build → Test → Learn
- Software: Design → Build → Test → Learn
- Modelling: Design → Build → Test → Learn
-
Go through at least one iteration of the engineering design cycle:
- Human Practices
Gold
- Wiki Writing
-
Special Award Nomination
- Best Software Tool
- Best Hardware
- Best Model
Design
Why these projects?
Projects Overview
Hardware
| Component | Priority | Objective | Assigned to | Report to |
|---|---|---|---|---|
| Microfluidic Pump | 1 | TBD | Piyush | |
| Microfluidic Chip (DNA Synthesis) | 2 | Create a chip with exposed -OH groups with highly turbulent flow over the mixing region to achieve Solid Phase DNA Synthesis. | Matthias, Patt | Tina, Piyush |
| Microfluidic Chip (PCR) | 2 | Create a connected PCR chip which can amplify free floating single stranded DNA. | Samuel, Piyush | Tina, Piyush |
| Microfluidic Chip (DNA Capture) | 2 | TBD | Samuel, Piyush | Tina, Piyush |
| Bioreactor | 2 | Create a Bioreactor to allow Wet Lab to Culture E Coli for their experiments | Samuel, Piyush | Tina, Piyush |
Software
| Component | Priority | Objective | Assigned to | Report to |
|---|---|---|---|---|
| Encoding | 2 | Implement a software pipeline that when given a file location to write to DNA, creates the DNA sequence(s) to be synthesized | Sebastian, Lucy | Lucy |
| Decoding | 2 | Implement a software pipeline that when given a request to retrieve a file, does the opposite of encoding | Riya, Lucy | Lucy |
| In silico testing | 1 | Given an input string, error rate of DNA synthesis generate faulty sequences to test against our encoding/decoding algorithm | Lucy, Sebastian, Riya | Lucy |
| Error Correction | 1 | Create an error correction algorithm to work with semi-specific encoding, short nucleotide sequences and high rate of deletion errors. | Riya, Lucy | Lucy |
| GUI | 3 | Design a user friendly interface that can be used by our iHP interviewees to try out our DNA storage software | All software + other dry lab if desired | Lucy |
Modelling
| Component | Priority | Objective | Assigned to | Report to |
|---|---|---|---|---|
| TdT Reaction Kinetics | 3 | Design an expression for the reaction kinetics of TdT. The result would be an expression where we could find the concentration of cofactors, reagents, etc that allow the kinetics of nucleotide addition to be equal for all nucleotides (A, T, G, C) | Lucy, Piyush | Wet Lab Leads |
| Bioreactor Modelling | 3 | TBD |
Software
Contributions
Lucy, Riya, Sebastian, Samuel
Overview, Context and scope
This document is related to the encoding/decoding pipeline that will convert binary information into nucleotides for the DNA storage process.
Some terminology that will be mentioned across the software pages include:
- frontend: portion of software that a user directly interacts with, another word for GUI
- GUI: graphical user interface
- backend: portion of software that the user does not see and interact with directly. Comprises of the encoding, decoding and error correction algorithms.
What does software want to try and achieve?
- In silico: Demonstrate ability to encode and decode information someone may store in long-term storage, in the 1000s of nucleotides long.
- Wet lab: Demonstrate ability to encode and decode a 100 nucleotide sequence with 30% error.
DBTL Cycles
Iteration 1 (Proof of Concept)
Implement a barebones pipeline, and see how much error can be tolerated in 100 nucleotide long DNA sequences with in silico testing.
Encoding
Given an existing file, convert that file into sequence(s) of nucleotides.
- Primer generation
- Sequence generation (semi-specific and specific)
Decoding
Given the name of a file that has been stored in DNA, decode that file back to binary information
- Return the primers to wet lab needed to retrieve the DNA molecules containing that file
- After returning the sequences, perform sequence recovery (based on the sequencing machine)
- Apply error correction based on synthesis strategy
- Collapse sequences back into one file
- Return the file to the user
Error Correction
Perform error detection and correction based on the synthesis strategy
ChaosDNA (provide platform for in-silico testing of software):
- Given a string, total error rate, deletion error rate, mutation error rate, insertion error rate, generate a distribution of sequences that may be synthesized from wet lab
- Run our software on these faulty sequences
- Run statistical analysis on different encoding and error correction strategies
Iteration 2
Redefine algorithms to tolerate up to 30% error in 100 nucleotide long DNA sequences, with in silico testing.
Iteration 3
Implement DNA Storage Alliance specifications, and do in silico testing on DNA sequences with 1000s of nucleotides.
Iteration 4/5
Test our software on sequences synthesized by wet lab, and redefine algorithms with in silico testing and wet lab data.
How do we test this?
The most important portion of our backend to test is the error correction. To complete iterations of the E-DBTL cycle, testing in silico will occur. Inspired by chaos engineering, we will create ChaosDNA, a simple tool that simulates faults (deletions, insertions, mutations) in DNA sequences. Using ChaosDNA, we can alter the rate of total error, then alter the proportions of deletion, insertions, mutations. We then run the error correction mechanism can against the faulty strands.
When wet lab has finished their POC, we can give wet lab different sequences of varying levels of redundancy to see the effect of adding more error correction and try to identify the most common type of error that occurs.
For primer generation, we can use open source tools online to test our primers, and later verification from the wet lab.
To test the graphical user interface, we will conduct interviews with the general public to see how they would use our tool.
Primer Generation and Sequence Generation
Primer Generation
Contributions: Lucy, QingRu, Achint, Tina
Why do we need primers?
Primers are important for DNA synthesis in our bodies. Usually 5-22 nucleotides long, primers are ssDNA that serve to “prime” or prepare a template strand for an enzyme to bind and initiate DNA synthesis. We will be generating primers with the four bases of DNA, because they are easier to synthesize and more stable than RNA based primers. Unlike DNA polymerase, TdT is unique, and does not require a template strand, so we will be focussing on generating primers that TdT can bind to and initiate DNA synthesis.
Storage-wise, primers act as unique identifiers for the data that is encoded in the information portion of the DNA sequence. Software-wise, the only strict requirement is that it is easy to generate unique primers; however there are biological constraints we must adhere to.
We want to generate primers that confine to these constraints, with the goal of ultimately generating acceptable primers for wet lab to order. Given a set of requirements, create primers that the wet lab can use for synthesizing ssDNA with TdT. Additionally, demonstrate that our data storage model will not run out of primers, and that we can create more primers to append/make edits to preexisting files [@Sharma_Lim_Lin_Pote_Jevdjic_2023,].
How are we generating primers?
Primers will be generated using a “genetic algorithm” [@Wu_Lee_Wu_Shiue_2004]. This requires:
- initial set of k primers
- fitness function
Successors, which are children of the "initial set" of primers, are generated by:
- Selection of two parents by random sampling, as determined by fitness function
- Crossover
- Mutation
These children are then checked against a set of constraints. If these constraints are satisfied, these children primers can be used, otherwise, these children primers become new parents.
This cycle can continue for as many iterations as we want A fitness function is determined by constraints, each having a weight or “acceptable” range
- primers that fulfill less constraints will have low fitness or be outright rejected
Some constraints include:
- determining melting temperature [@addgene2019]
- determining if secondary structures will form, simple reverse string check is performed, but more advanced checks will be tried in futher iterations [@primerpcr].
How do we test this?
We can use open source tools that evaluate the melting/annealing temperature, secondary structure formation and other constraints to ensure our code is generating acceptable primers. We can also verify by their use in wet lab, however this may not be feasible given lack of time and resources.
Sequence Generation
Contributions: Lucy, Riya, Sebastian
How do computers interpret bits?
Given a user’s file, we must convert that file, which contains many bits, to a collection of approximately sized nucleotide sequences for synthesis by wet lab. First off, what is are bits?
A bit, either 0 or 1, is the most basic form of information a classical computer can interpret, meaning data that is stored and interpreted on a computer is in the form of 0 and 1.
A set of bits can have any meaning if there is no context provided behind how to decode these bits. We can interpret bits as a number, or a character.
- if we interpreted these bits using UFT-8: 00100100 -> $
- if we interpreted these bits as a number: 00100100 -> 36
The mapping from binary sequences to characters is standardized via the UTF-8 (Unicode Transformation Format - 8 bits) standard [@wikipediautf].
UTF-8 is a standardized format for storing and reading characters. UTF-8 encodes for characters, symbols, etc. There are other standards, such as ASCII, which are cover less characters than UTF-8. For instance,
- 11100000 10100100 10111001
- using UTF-8: ह
- using ASCII: ह
When we encode information, it is important to note down, either on the actual DNA strand itself or on a computer, how to interpret these bits once we read back the DNA strand. This is what is called metadata, data that tells you information about data!
How are bits converted to nucleotides?
There are several ways to change a bit sequence into a DNA strand, and a few are listed below:
- base4 encoding: 0 -> A, 1 -> T, 2 -> G, 3 -> C
- Church encoding: 0 -> A or C, 1 -> G or T
- base2 encoding: 00 -> A, 11 -> T, 01 -> G, 10 -> C
- HEDGES encoding
- Rotation based cipher
We will be implementing the HEDGES encoding and rotation based cipher.
What is the rotation based cipher?
[@Bornholt_Lopez_Carmean_Ceze_Seelig_Strauss_2016]
Because TdT only adds semi-specifically, meaning it adds bases until it runs out of bases, we can select which base TdT should synthesize, but not how many (there are ways to get around this, but they take more time to implement). Thus, using a rotation based cipher, we can get around this peculiar behaviour of TdT.
The transitions in bases encode for 0, 1 and 2. This cipher encodes for information in base3, whereas computers usually interpret information in base2. We can get around this by converting base2 information to base3. Then, we must select an arbitrary start base, and then follow the arrows to encode information.
What is the HEDGES encoding?
HEDGES is a type of encoding similar to fountain codes, which tries to encode redundancy using a hash function [@press_2020_hedges]. HEDGES is a type of key-autokey cipher, which means it incorporates portions of the message (our bit sequence) into the encoded nucleotide sequence. Again, what does this mean?
This means for a bit in position i that we want to encode as a nucleotide, we must use the bit sequence up to the position i we want to encode to generate the next nucleotide. The most important thing to understand that a hash function, which takes as input a stream of bits, is used to create the nucleotide sequence.
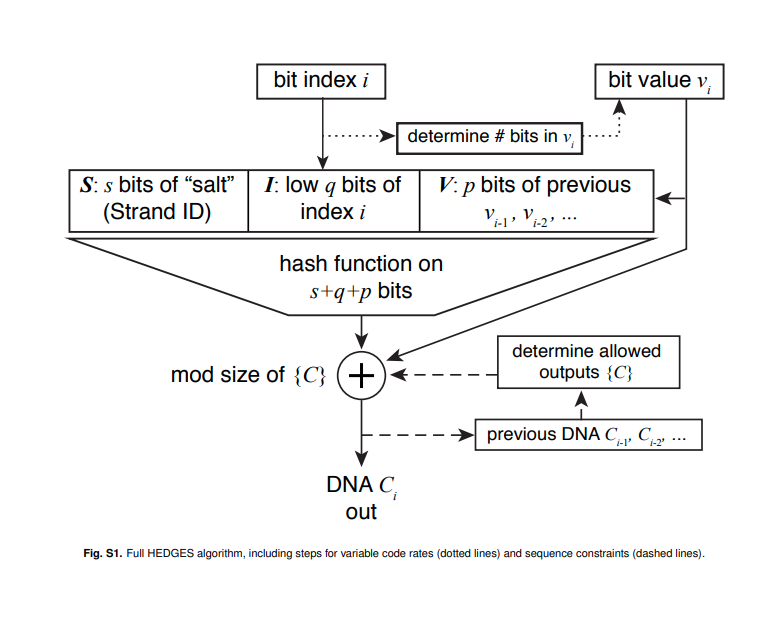
What is an high level overview of sequence generation?
- File format
- the file format tells us how to interpret bits that encode that file
- without knowing the file format (the file extension), we have no idea what the bits encode for
- Compression
- if there is no special compression strategy, we will just compress using a generic compression algorithm, otherwise, we can take advantage of certain file formats and compress in a special way
- Block the bit sequence
- we break the information into chunks of around 80 - 100 nts long
- if we are doing semi-specific synthesis, probably around 20 - 30 nts long
- Implementing encoding strategy (convert bit sequence to nucleotide sequence)
- if we are doing specific synthesis, we will encode the binary information according to HEDGES
- if we are dong semi-specific synthesis, we must use the rotation based cipher
- Add outer codes
- a form of purely redundant error correction, only possible if we are doing specific synthesis
- Collect metadata
- Give sequences to wet lab
How do we test this?
The robustness of our redundancy, collection of metadata will be evaluated
- In silico: with software generated faulty DNA sequences
- In lab: but probably not enough times to be statistically significant
File compression and decompression
Primary goal
By encoding a heavily compressed file, we can effectively increase the amount of information stored in DNA for a given number of nucleotide bases.
Key points
- Data compression: defined in information theory as the process of encoding information using fewer bits than the original representation. In the context of our work, decreasing the number of nucleotide bases required to encode a given file.
- Token (LLMs): the fundamental data unit within natural language processing systems such as large language models (LLMs). Most common AI systems used today are some form of LLM (e.g., ChatGPT, Google Gemini, Diffusion-based models such as Stable Diffusion). A token essentially acts as a small component of a large data set; when an LLM takes text input, such as a sentence inputted into a chatbot, it breaks the query down into a set of tokens. These tokens are then processed by the model.
- Lossless compression: a compression process that does not result in any data loss.
- Lossy compression: a compression process that results in data loss. For instance, when audio is compressed into common file formats such as .mp3, audio quality is sacrificed to decrease file sizes.
- Compression ratio: the ratio between the file size of the inputted and outputted files. Often expressed in bits per base (bpb, output/input).
Text compression
Dictionary compression (traditional)
Dictionary compression is a traditional text compression mechanism where a "dictionary" is constructed with words or phrases that are commonly used. For example, if the phrase "How are you?" is repeated many times throughout a text, we could define "How are you?" = 0110011001110001, a binary sequence with 16 bits. Thus, we can replace every instance of "How are you?" with this 16-bit identifier as opposed to the original text string which is 96 bits long.
However, dictionary compression functions best when a new dictionary is created for every document inputted, and does not always result in a high compression ratio (especially if repeats are not extremely common in the text).
Tokenization and the ts_zip utility
ts_zip is a small utility that enables text compression through tokenization. An inputted text is broken down into tokens, and the token "values" are saved as a binary file. The binary file can then run through the same process in reverse; each token is converted back to the string that it is related to. As long as the same model is used for compression and decompression, this process is lossless. In fact, the process is relatively similar to dictionary compression, except the LLM model is used as a static dictionary for all input files, and the token values are used as identifiers.
Thus, the model used for compression must be careful selected, with a focus on optimising for model size, compression speed, and compression ratio. A lower compression ratio (greater efficiency) and a shorter compression time is ideal. Thus, four models - falcon_40B, rwkv_7B, mistral_7B, and gptneox_20B - were evaluated for their relative performance. The benchmark results and technical specifications are shown below.
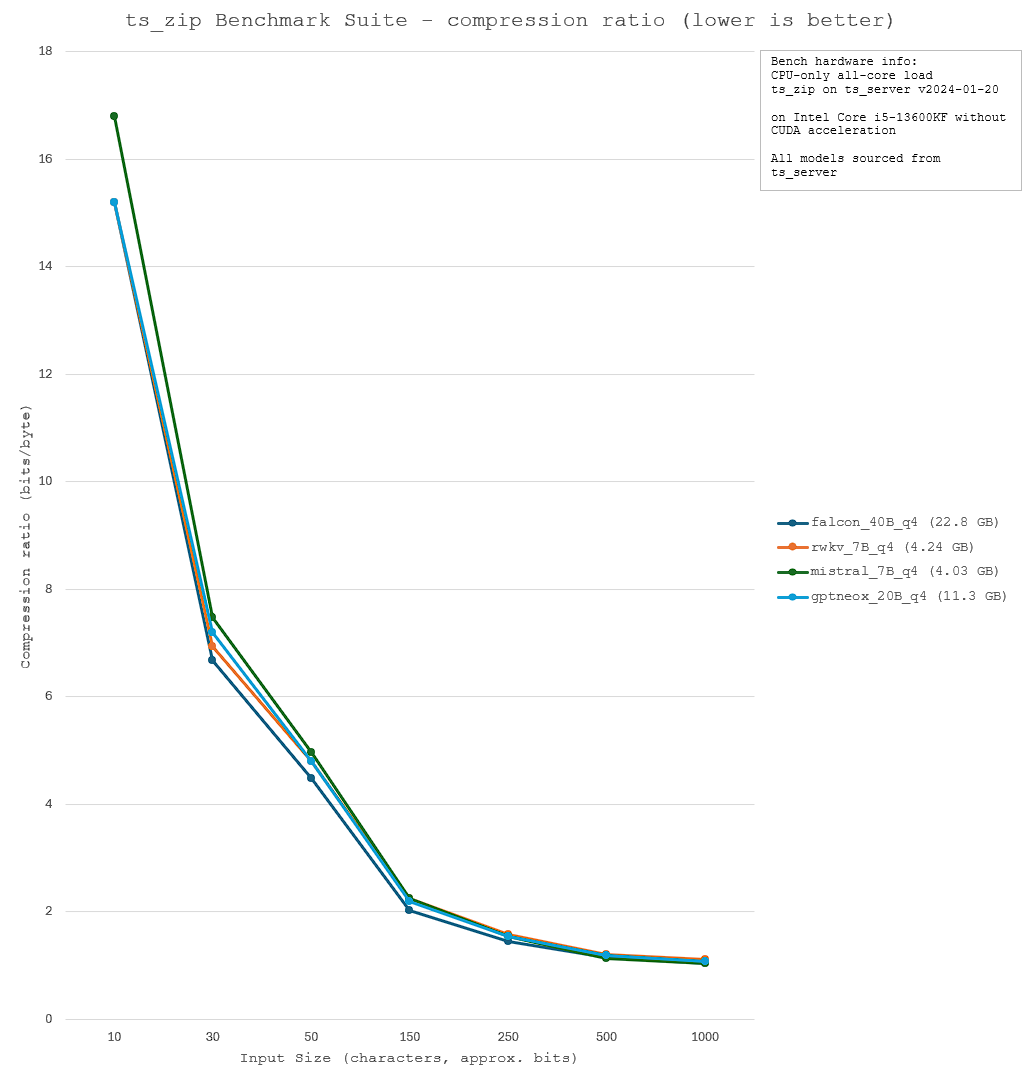
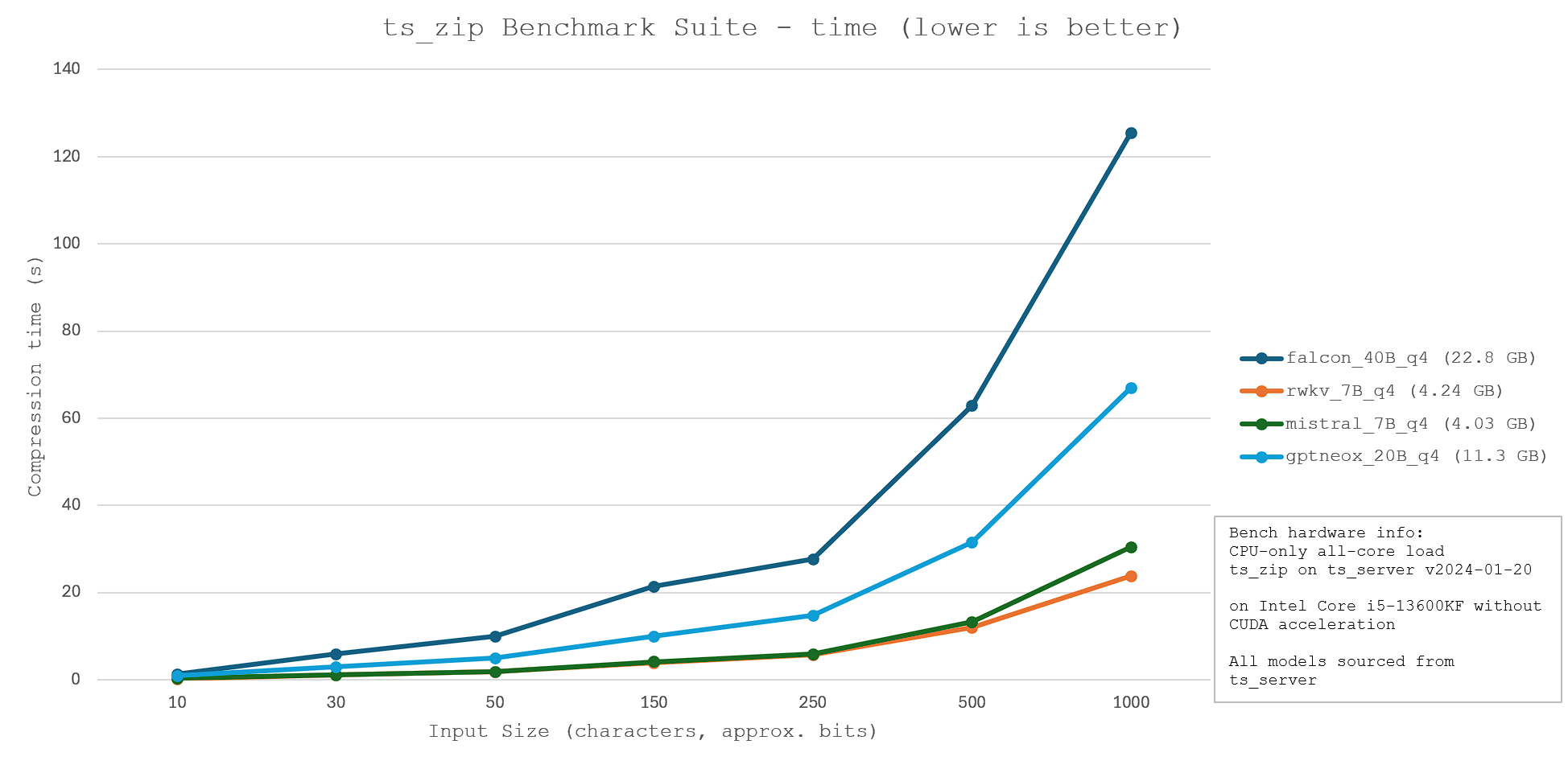
Other compression algorithms:
GZip: https://www.gnu.org/software/gzip/
Gzip is a commonly used compression algorithm and filetype. It can be used to compress many different source filetypes, but is non-competitive compared to ts_zip in terms of compression ratio for text files specifically. However, should be considered for svg compression.
Bzip2: https://en.wikipedia.org/wiki/Bzip2
Bzip2 is similar to Gzip; a compression algorithm with diverse use cases. Typically slightly worse-performing than Gzip.
LZ4: https://github.com/lz4/lz4
Unique compression ratio that offers modes for higher compression or faster speeds. Requires more complex installation for use; has numerous dependencies. Potentially competitive with GZip for certain use cases. Currently not benchmarked; will be further investigated.
Error Correction
Contribtions: Lucy, Riya
- Overview, Context and Scope
- How does synthesis method affect error correction?
- Semi-specific synthesis
- Specific synthesis:
- Current solutions
- How do we test this?
Overview, Context and Scope
Correcting for errors that occur in the DNA synthesis, storage, sequencing process, as relating to our DNA synthesis method. TdT, and DNA synthesis and sequencing in general have very high rate of error.
A benchmark for the percentage of errors we may be dealing with:
- % Reads containing errors [@lee_2020_photondirected]:
- Single base deletions: 25.8%
- Single base insertions: 13.4%
- mismatches: 8.9%
We must collect enough metadata to correct a DNA sequence with around 30-50% synthesis error [@_2021_teamaachenresults]. We should avoid adding error correction bits unless there is drastic improvement. This could depend on the type of data we are encoding. To complete an iteration of the DBTL cycle, we will implement a simple error correction algorithm for both semi-specific and specific synthesis, followed by testing in silico. Based on the results of the algorithm, we either enhance it or abandon and try another algorithm.
How does synthesis method affect error correction?
The synthesis method differentiates our sequence recovery method. If we are using semi-specific, we can rely on homonucleotides and number of transitions for sequence consensus. For specific synthesis, we would rely on error correcting codes.
Semi-specific synthesis
Given that our synthesis method is semi-specific: this means we can control the type of base we are adding, but we cannot control the number of bases. Additionally, because we will be attaching “blocks” of bases, such as “AAAA” when we just want “A”, a nucleotide sequence of 100 bases may only contain 20-30 unique nucleotides
The challenge here is how can we apply error correction:
- With a limited number of unique nucleotides per strand
- Where deletion errors are the most likely to occur
- Where sequences are short (100 nt)
- To be robust such than it an tolerate higher rates of error
What metadata should we collect for semi-specific synthesis?
The number of transitions will be collected, which we can see if it is enough metadata to decode a faulty nucleotide sequence.
How is error correction done with this metadata?
Using metadata collected during encoding and checksum, systematically guess which base transitions occur. We first find how many base transitions are missing, and try insertions, deletions, mutations to match the recorded metadata. This is similar to solving sudoku, e are guessing which base transitions are correct. We will use stochastic estimation to choose the “most likely” correct bases. Everytime some “constraint” is violated, we can either backtrack or create a new “sudoku” to solve.
We try solving this problem for some allotted time; if the algorithm fails to return we mark the strand as too erroneous to recover and signal failure to the user, otherwise, we move onto to reconstruct the file.
Specific synthesis:
Given specific synthesis means we can control the type of base and how many of that base we add. However, gaining the ability to add specifically means deletion errors and insertion errors are more detrimental since there is no redundancy (unless we explicitly add it ourselves). The advantage is that we can choose to how we want to encode the redundancy. We will first try to encode redundancy using HEDGES [@press_2020_hedges].
Inner codes
Inner codes refers to bases that encode for redundancy and information. HEDGES is a type of inner code. For more on HEDGES, refer to encoding. If you are interested, I highly recommend you read the paper [@press_2020_hedges].
Outer codes
Outer codes only encode for redundancy, but can be more powerful than inner codes. A type of outer code is reed solomon codes (with GC++) [@hanna_2024_short], which we will also implement.
How do we decode a HEDGES encoding nucleotide sequence?
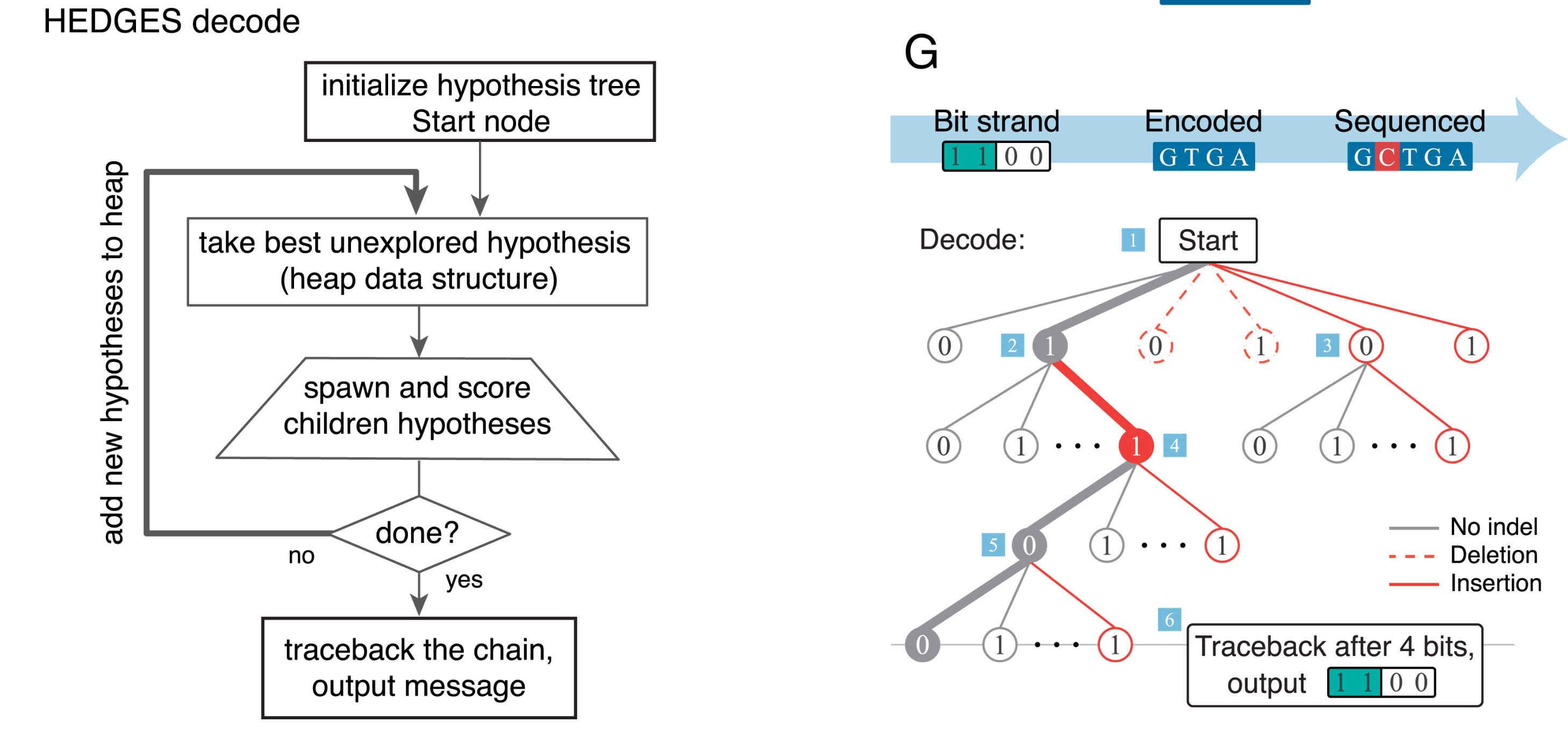
Read the HEDGS paper if you want to know more [@press_2020_hedges].
Current solutions
For more on these papers check out
- HEDGES: Hash Encoded, Decoded by Greedy Exhaustive Search [@press_2020_hedges]
- To reduce the number of ECC bits needed to be encoded, some store of probabilistic estimation of the next base must occur
- “physical sequence redundancies, a stringent filtering process and stochastic estimation” [@lee_2020_photondirected]
- https://github.com/dwiegand740/Photon_Enzymatic_Synthesis
- Stochastic estimation via matlab’s built in “seqlogo”: https://www.mathworks.com/help/bioinfo/ref/seqlogo.html
- GC+
- Short Systematic Codes for Correcting Random Edit Errors in DNA Storage: https://arxiv.org/abs/2402.01244
- QR Code error correction:
- Naive Redundancy
- "To overcome the challenge of correcting deletions and insertions, existing methods often rely on deep sequencing, which generates many reads per oligo. This injects sequencing redundancy analogous to repetition coding, typically leveraged through sequence alignment algorithms to correct edits via majority voting." [@hanna_2024_short]
How do we test this?
- We should see if added ECC bits actually increase the accuracy of information; need to perform statistical analysis
- Or is the actual sequence more important
- Via ChaosDNA
Decoding
- Overview, Context and Scope
- What types of sequencing machines are there?
- What algorithms are we using?
- Why do we need to remove primer from the sequence before we do error correction?
- Sequence collapse to single nucleotides
- Current Solutions
- How do we test this?
Contributions: Lucy, Chae, Riya, Sebastian
Overview, Context and Scope
After DNA is sequenced, we must perform some work on the sequenced strands before we can decode the DNA. This includes alignment of fragments of DNA sequences after sequencing, and the methodology depends the sequencing platform we use.
Many pre-existing alignment strategies are used in the realm of bioinformatics, and rely on a reference template; we don’t have that luxury and conduct sequence alignment without a template, otherwise known as de novo sequence assembly.
To perform iterations of the DBTL cycle, we will complete the algorithms required to perform alignment in a timely manner and with acceptable accuracy on one platform, most likely NGS. We will implement one of the two algorithms that is mentioned below, then in the second iteration try the other algorithm.
What types of sequencing machines are there?
Sanger
- [@a2021_analyzing] [@a2021_sanger]
- First 20-40 base pairs are not well resolved
- Simple data analysis
- Longer reads (500-700 bps)
- Low sensitivity (~15–20% detection limit)
- Sequences close to primer-binding sites to be of poor quality
- Output: four-color chromatogram representing the peak fluorescence intensity associated with each labeled ddNTP along the DNA sequence
NextGen
- [@next] [@cheng_2023_methods]
- Higher sequencing depth for increased sensitivity (down to 1%)
- Higher discovery power
- Short reads (150-300 bps) by Illumina
Nanopore
Doesn't require PCR amplification, eliminating amplification bias and simplifying sequencing protocols relatively high error rates, around 10% per nt [@emerging].
What algorithms are we using?
There are many established algorithms in this domain, we will use one of those. Based on the selected platform, sequence alignment will be performed as so:
Sanger
Given four-color chromatogram(s) representing the peak fluorescence intensity, depending on the amplification scale in wet lab, we can compare chromatogram(s) to each other to resolve conflicts. For de novo assembly, we can resolve conflicts based on redundancy of chromatograms [@sanger].
NGS
A fastq file contains multiple chopped up sequence reads, each having a confidence score known as a Phred score. A Phred score is the probability the sequencer called the base incorrectly. We assemble the sequence reads de novo into one long read based on the overlaps between the chopped up sequences [@ngs].
To conduct de novo assembly for output from NGS platform, reads are examined for overlap between them, and the goal to build up a single contig from smaller contigs.
There are established algorithms for solving this problem [@paszkiewicz_2010_de]:
The overlap-layout-consensus (OLC) approach [@paszkiewicz_2010_de]
- Find overlaps between all pairs of segments, deriving a similarity score for each pair; this will be used to generate our heuristic
- Then we have to generate the layout based on overlaps, with an overlap graph
- Vertices: sequence reads
- Edges: overlaps
- Find a path that visits each node once -> Hamiltonian circuit, NP-hard
- So use heuristic (similar score) to greedily select which edges to take to maximize the heuristic (sum of similarities) until a single string is found (path is found)
- Order of merging pairs matters and can change the final string
Requires overlap to be scored between all pairs of reads, making runtime as least O(n2), overlap is less likely for short reads.
The de Bruijn graph approach [@paszkiewicz_2010_de]
- No need for overlap phase
- Sequence reads cut into smaller pieces
- K-tuples, DNA sequence word of length k, are generated
- Bruijn graph:
- Node: (k-1)-tuples that occur in k-tuples
- Edge: connects (k-1)-tuples that form a k-tuple
- Find shortest (minimum weight, using heuristic) path that visits every edge -> Chinese postman problem, NP-complete! ** Problems with finding multiple solutions, or sequencing errors which cause high branching and tangle
- Effect choice of k
Nanopore
TBD, as we may not end up using Nanopore to sequence.
Why do we need to remove primer from the sequence before we do error correction?
We have to remove the nucleotides representing the primer in order to only decode on the nucleotide strand that contains information bases. Using the primer we have stored, we can run fuzzy string matching algorithms [@wikipediacontributors_2019_approximate]. We must use fuzzy (approximate) algorithms because there is a chance the primer doesn't quite match the primer sequence we have stored (due to errors in synthesis and sequencing).
From simplest to advanced [@silva_2022_what]:
- Levenshtein distance: used in strings
- Damerau-Levenshtein: “transposition of two characters to find an approximate match”
- Hamming distance: used in signals
- Advanced: Hidden Markov Models via probability
For the first iteration of the DBTL cycle, we will try Levenshtein Distance, and pursue other algorithms if there is notable gain from using them.
Sequence collapse to single nucleotides
Given that we are doing semi-specific synthesis, we now collapse homonucleotides to mononucleotides. We use the occurrence of homonucleotides as the probability the sequenced base is actually at that index, to deal with base conflicts, and also signal that the positions of base conflict could either be a deletion, insertion or mutation error.
After these steps, we can do error correction.
Current Solutions
- Basecalling, alignment, assembly and deconvolution of Sanger Chromatogram trace files
- Aachen
- Open source algorithms for short reads:
- “These software packages are able to perform de novo assembly of Illumina short sequence reads with the exception of SHORTY, which is designed to assemble ABI SOLiD colour-space data. Velvet and SOPRA can assemble sequence-space and colour-space data. aCurtain is a pipeline, based on Velvet, for hierarchical assembly of short sequence reads in order to overcome memory usage limitations. bOases is specifically designed for assembling transcribed sequences.” [@paszkiewicz_2010_de]
How do we test this?
We can test in silico by using open source genome data, and try to reassemble (without the reference template) and then check the performance, additionally through ChaosDNA.
ChaosDNA
Context and Scope
To perform E-DBTL cycles without data from wet lab, we can generate faulty DNA sequences through software. Treating DNA sequences as a string, we can randomly mutate the string with deletions, insertions and mutations.
Goals
The goal of this in-silico testing platform is to perform 3-4 E-DBTL cycles before wet lab has data for us to try. Additionally, because wet lab will only be generating strands of 100 nucleotides long, we want to try our software on nucleotide sequences that are 1000s of bases long, and run statistics to show the utility of our software with input sizes that will be more realistic of information that would be encoded in long-term storage.
Design
- To test sequence generation, error correction: given a file to encode, total error rate, deletion error rate, insertion error rate, mutation error rate, return a distribution of faulty DNA sequences
- To test our sequence alignment (NGS): return a DNA sequence in the form of a fastq file
Enzyme Kinetics
Overview
To create a model of DNA synthesis with terminal deoxynucleotidyl transferase (TdT), where parameters such as concentration of reagents can be optimized to ensure kinetics of nucleotide addition are equal to each other [@a2023_terminal].
Context and Scope
TdT adds bases promiscuously, meaning the four bases of DNA do not add at the same rate [@motea_2010_terminal]. This makes semi-specific synthesis challenging, as under conditions where concentrations of reagents are not tailored to each nucleotide, each of the four different nucleotides will add at different rates, meaning the number of nucleotides added will be different; we may add the A nucleotides very well, but struggle to add one single T nucleotide. This was a problem faced by another iGEM team, resulting in a high rate of deletion of bases.
Cofactors
TdT uses divalent metal ions to facilitate addition of nucleotides. The concentration and type of divalent metal ion will probably have the biggest impact on the rate of nucleotide addition. Divalent metal ions that TdT can use include Co2+, Mn2+, Zn2+ and Mg2+.
- General notes [@motea_2010_terminal]:
-
Extension rate of dATP: Mg2+ > Zn2+ > Co2+ > Mn2+
-
Mg2+: prefers dGTP and dATP
-
Co2+: prefers dCTP and dTTP
-
Zn2+: positive effector for TdT, when micromolar quantities are added with Mg2+
- Induce conformational changes
-
Polymerization rates lower in presence of Mn2+
-
The concentration of metals ions will be important to model, as “metal ions directly influence the mechanism of template-independent polymerization by changing the location of the rate-limiting step” [@motea_2010_terminal].
Temperature
WT TdT and thermostable TdT synthesize DNA at different temperatures [@puay_2020_evolving]. This means WT TdT modeling may not accurately reflect the kinetic capacity of thermostable TdT.
Nucleotide types
If we want to add fluorescent nucleotides, it is not certain if these nucleotides with an extra benzene/organic molecule add at the same rate as WT nucleotides, since the ultimate goal is to synthesis DNA with wild type nucleotides. Literature has stated that “TdT uses non-natural nucleotides such as 5-NITP with identical efficiencies as dGTP, the preferred natural nucleotide.” [@motea_2010_terminal], however, TdT has "inability to elongate bulky non-natural nucleotides may be due to steric constraints from 16-amino acid loop" [@motea_2010_terminal].
Goals and non-goals
- Design a model of WT TdT enzyme kinetics to ensure rate of base addition is equal for each base, and transfer that model to thermostable TdT.
The actual design
Parameters to model/vary
- Cofactors
- Nucleotide concentration
- Enzyme concentration
Parameters to collect?
- Concentration of varying cofactors
- Concentration of enzyme
- Initial rate:
- Require Early linear potion of Concentration vs. time profile
- RPKA/VTNA:
- Two or more time course reaction profiles compared in pairwise function, initial concentration of reagents/catalysts varied to determine impact
Model type
The model type depends on the TdT mechanism; TdT functions via “rapid equilibrium random kinetic mechanism”, and “TdT catalyzes DNA synthesis in a strictly distributive mode” [@motea_2010_terminal].
Michaelis-menten Model
According to Aachen, this model, for single substrate reactions, is not suitable according to Aachen, since DNA strand is not always attached to enzyme [@_2021_teamaachenmodel]. So we must look for another model, such as a multi substrate model [@a2019_56].
Multi substrate model
If there are multiple substrates, they can bind sequentially or non-sequentially. The order of binding also matters, where in an ordered reactions, one substrate must bind before the other. Experiments are designed to keep substrate A kept constant, while substrate B is varied, to make the problem, According to Aachen TdT is two substrate, two product random order mechanism [@_2021_teamaachenmodel].
General guide to reaction optimization [@deem_2023_best]:
- Find initial reaction conditions, which can be identified through small-scale batch end point screening. This will probably be done with gel, but for sake of time we might be forced to look into literature.
- Initial time course survey, a fast/quick way to get an initial understanding of the reaction at hand. Identify if there is induction period, to see what assumptions we can form, for instance if the reaction is under steady state.
- Reoptimization, using initial time course survey
- Control experiments, to see effect of order of addition, change in sample composition over time and whether the enzyme is stable over duration time.
- Reproducibility, to show reproducible workflow with final reaction conditions collected in step 3.
- Validate method of quantification; how is collected data converted to concentration vs time?
- Kinetic experiments, which can we run after above is done, use RPKA and VTNA to establish kinetic relationships.
- Validate conclusions, with a hypothesis, usually reaction modeling done in silico via Dynochem, COPASI, Zenth.
Real time monitoring?
For more details, refer to wet lab documentation. Monitoring of DNA length is going to be done through gel (not real time, can't tell which base is being added) or fluorescence (requires modified nucleotides).
Gel
- Transfer reaction into gel and run it (~20ul) → run gel → can see different lengths (mass)
- Can’t check specific bases that were added
- Want to test if it can successfully add anything given a set rxn parameters
Single-Molecule Fluorescence Microscopy:
- Benzo-expanded dxNTPs [@jarchowchoy_2010_fluorescent]: Up to 30 consecutive monomers can be incorporated by TdT
- Mixture of labelled dUTP and dNTPs
- Mixture of fluorescent dNTPs and natural dNTPs
Current solutions
Aachen used two substrate model instead of MM. Additionally, modelled DNA pools, where for each length of DNA strand, new pool created, flux from one pool to another modeled. They found that concentration of DNA strands with no elongation lead to error [@_2021_teamaachenmodel].
- Code: https://static.igem.org/mediawiki/2021/5/5f/T--Aachen--documents--Modeling--modeling_code_igem_aachen_2021.zip
- We could reuse their code, refit to our parameters, and directly start testing
How do we test this?
- Dry lab can start creating the model in silico and run the reactions in silico, and then use wet lab data as it becomes available.
How long will this take?
From whenever dry lab begins modeling, to the end of the wet lab experiments.
Microfluidics
Overview
Microfluidics is a technology that involves the precise manipulation of fluids at the microscale, often within channels of tens to hundreds of micrometers in size. This technology is particularly significant in biomedical research and diagnostics due to its ability to handle small volumes of fluids with high precision and control, leading to reduced reagent use and faster analysis times [@origins_microfluidics].
In our DNA synthesis project, microfluidics can revolutionize the process by enabling the rapid and efficient assembly of DNA sequences by facilitating the precise delivery and mixing of nucleotides in controlled conditions. This allows for the synthesis of long strands of DNA with high accuracy in a platform that can support high-throughput synthesis through parallel production of multiple DNA sequences [@outlook_microfludics].
Context and Scope
In the context of our overall DNA synthesis topic, microfluidics will be taking on 3 major projects:
Project 1: PDMS/PMMA-Glass DNA Synthesis Chip
The easiest chip to produce given Dr. Following Folch's advice, this chip would involve vertical and horizontal channels cut into PMMA, allowing reagent to flow over a glass channel. DNA synthesis would occur over the glass section, using techniques and reagents supplied by wet lab.
Project 2: Fluid Distribution System
Here we design a microfluidic pump and reservoir system that can take reagents up from separated wells, mix them macroscopically or in a microfluidic mixer, and then circulate them through a microfluidic chip before recovering them into a secondary reservoir.
Current ideas for the pump include a peristaltic, or syringe based negative pressure pump. Fluids could be redirected macroscopically using 3D printed valves, examples of which exist on Arduino forums. Closed circuit circulation is currently, and will likely need to be solved macroscopically as well.
Project 3: PCR on a Chip
Once we have ssDNA, the same chip’s reaction chambers would be responsible for converting it into dsDNA, before supplying it to a PCR on a Chip module. Here, we would attempt to amplify the generated DNA, to ensure more copies being present before going into long term storage. This last project is very ambitious, and likely time and skill dependent. It will depend on a number of factors, primarily Samuel and Piyuh’s motivation and availability during the summer, and access to the SBME makerspace.
Goals and non-goals
At the very least, it is imperative we finish Project 1. It is no longer technically challenging and is necessary for our project to be holistic - and it would be a requirement for our Gold Medal.
Project 2 would add a layer of “splendor” to the process, but is not necessary for the functionality of the process. It would help us with our petition for a project award, past a medal requirement.
Project 3 is very impressive, as it has only existed in literature and never been attempted in this timeframe before. It would require substantial makerspace time and near constant testing, but if pulled off would guarantee some sort of award.
Diagram
The two for the DNA synthesis design are as follows:
Here, the channels are made out of PMMA, and are mostly lateral. This would allow us to utilize a laser cutter in the Makerspace and not require any additional materials, although the BioMEMs laser cutters could be more useful due to a higher resolution.
For our valves, look at this to get a good idea of what we want to do:
https://www.instructables.com/3d-Printing-Servo-Controlled-and-Other-Valves/
For our project, we will likely be using a combination of pinch valves and check valves.
Finally for the pump, we would either be going with a 3D printed peristaltic pump, or a syringe based negative pressure pump. Check these links out for examples:
https://blog.arduino.cc/2023/02/22/a-diy-peristaltic-pump-controlled-by-an-arduino/
https://www.youtube.com/watch?v=0KjniIBHNMo
Build Process
The build process would follow the DBT cycle, and we would be documenting heavily using DHFs (since all BMEG is familiar with this, Samuel will be caught up to speed).
The process would begin with designing on SolidWorks, before 3D printing in the SBME Makerspace and assembly. We would be using Arduinos for all our electronic work. Some of the tools being used would be the laser cutters, soldering stations, drill press and plastic welding applications.
For microfluidic channels, we would need an additional step of flow simulation on COMSOL, before the fabrication step.
How long will this take?
- Project 1 should be complete by Early June.
- Project 2 should be complete by end of July.
- Project 3 should be complete by end of August.
Bioreactor
- Overview
- Goals and non-goals
- The actual design
- Diagram
- How do we test this?
- How long will this take?
Overview
A bioreactor is a device that provides a controlled environment for the cultivation of biological organisms (like bacteria, yeast, plant and animal cells) under specific conditions to promote growth. Controllable conditions include temperature, pH, oxygen levels, and nutrient supply, which are precisely manipulated to optimize the resident population’s biological processes [@bioprocess].
Developing a bioreactor early on in our competitive season significantly enhanced our project’s outcomes, by allowing for scalable production of W.T. E Coli for the numerous experiments that Wet-Lab aimed to complete. Additionally, it served as a training tool for rookie Wet-Lab members learning to culture E Coli., teaching them concepts of cell culture and molecular cloning through an error proof system.
Goals and non-goals
The motivation behind the Bioreactor was to provide Wet-Lab with a device facilitating rapid E Coli. cultures, allowing for a steady reservoir of host bacteria should there be a need for unplanned experiments. The decision to prioritize the construction of the first iteration of the Bioreactor was based on the team’s experience from the previous year, where leaving it to the end precluded the prototype from testing and implementation in Wet-Lab’s procedures. As such, development of the Bioreactor began in March 2024, and followed the Engineering design cycle through 4 iterations, until the final prototype was completed in mid-July.
The actual design
The Bioreactor iterated through 4 major design changes (denoted by Marks), with numerous modifications between each iteration (denoted by letters).
Bioreactor Mk. 1
This version of the bioreactor would have the following features AT MINIMUM:
- Stir Rod
- Passive Oxygen diffuser
The functionality is to just grow W.T. E Coli. somehow, and show it isn’t just in a culture. The stir road provides kinetic stimulation, the oxygen Diffuser maintains O2 levels via diffusion. I don’t expect a lot of iteration within this Mark, maybe 1 or 2 just adding dead spaces/module slots for later additions to the bioreactor. With the passive oxygen diffuser, E. coli can grow in both anaerobic and aerobic conditions so the diffuser is primarily responsible for regulating the pressure levels inside the reactor. If required, active diffusion and O2 readout can be added but are not necessary for efficient function.
Bioreactor Mk. 2
This version of the bioreactor would have the following features AT MINIMUM:
- Stir Rod
- Active Oxygen Diffuser (Pumps)
- Temperature Control Module
In Mk. 2, we add more variables to control the growth rate of the E Coli., and likely iterate within to optimize growth conditions. This would require significant wet lab experimentation, and we would attempt to reduce the experiment counts by using literature review to start off close to optimal conditions.
Bioreactor Mk. 3
This version of the bioreactor would have the following features AT MINIMUM:
- Stir Rod
- Active Oxygen Diffuser (Pumps)
- Temperature Control Module
- OD Readout Module
- O2 readout module
- Arduino Thermometer
Having found the optimal growth conditions, Mk.3 incorporates sensors for readouts. We would start with an OD sampler (for bacterial population) and O2 readout module (to verify oxygen saturation in the system) and an Arduino based thermometer that can send readouts digitally to a computer.
Bioreactor Mk. 4
This version of the bioreactor would have the following features AT MINIMUM:
- Stir Rod
- Active Oxygen Diffuser (Pumps)
- Temperature Control Module
- OD Readout Module
- O2 readout module
- Arduino Thermometer
- Bluetooth Module/WiFi Module
This would just be software updates. Using the same hardware, develop simple code to get real-time readouts while far away from the bioreactor. This would be great for monitoring purposes.
Diagram
Generic Diagram looks like this:
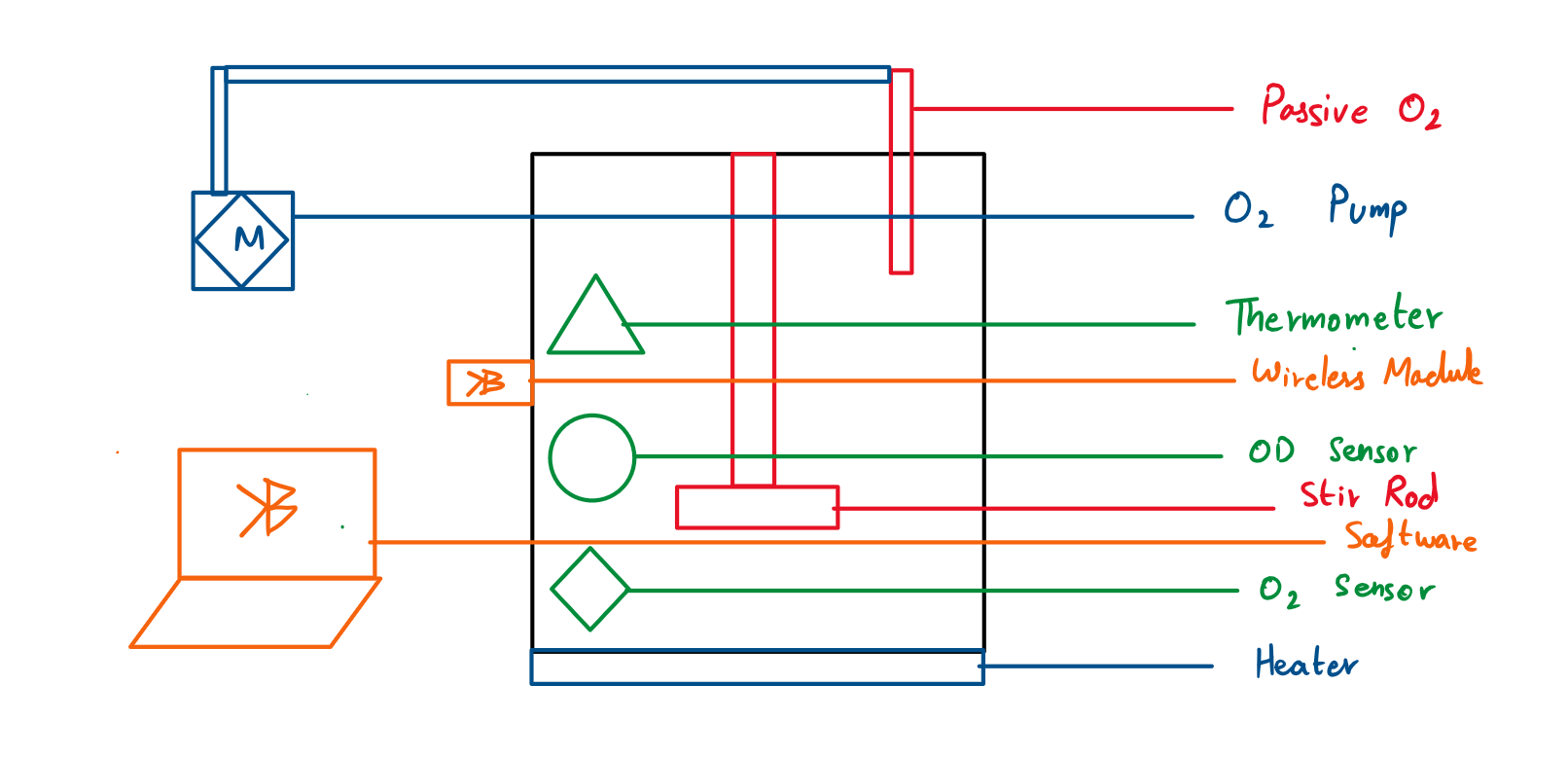
Red is Mk.1, Blue is Mk.2, Green is Mk.3 and Orange is Mk.4
How do we test this?
Testing will primarily be based on one metric: bacterial population in the bioreactor during midpoint of log phase growth.
How long will this take?
- It would be nice to have Mk.1 completed by mid-April
- It would be nice to have Mk. 2 be completed by end of May
- It would be nice to have Mk. 3 completed by end of June
- It would be nice to have Mk. 4 completed by mid September
Engineering Success
Software
Iteration 1 (March - April)
| Relevant Links | |
|---|---|
| Design | Software, https://github.com/UBC-iGEM/internal-wiki-2023-24/commit/1a63801e3c9fe8105b47d43dd3dccf7bc719fca8, Main Repo, ChaosDNA |
| Build | |
| Test | |
| Learn |
Iteration 2 (April - May)
| Relevant Links | |
|---|---|
| Design | |
| Build | |
| Test | |
| Learn |
Bioreactor
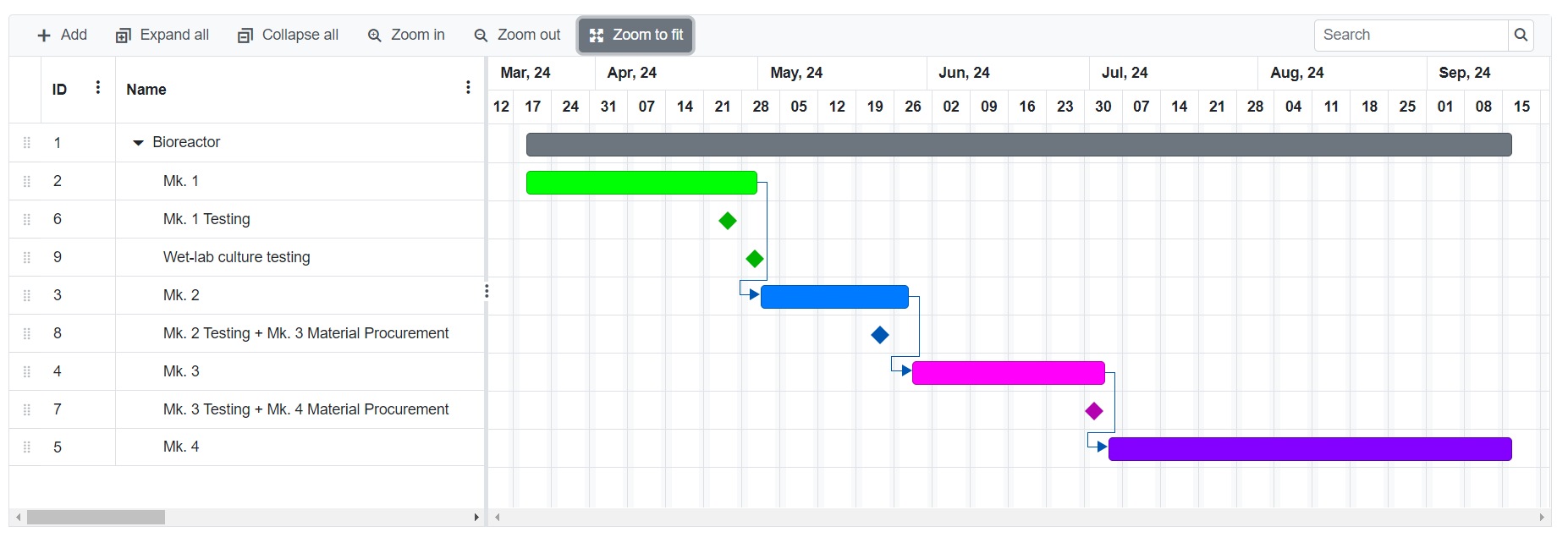
Iteration 1
| Relevant Links | |
|---|---|
| Design | Bioreactor, https://github.com/UBC-iGEM/internal-wiki-2023-24/commit/3b2b575853cb7d3921c12733e5c435574d70ca35 |
| Build | |
| Test | |
| Learn |
Microfluidics
Iteration 1
| Relevant Links | |
|---|---|
| Design | Microfluidics, https://github.com/UBC-iGEM/internal-wiki-2023-24/commit/3e90962005fea743407bb4eb9fa36a09b0b92b07 |
| Build | |
| Test | |
| Learn |
Modelling
Iteration 1
| Relevant Links | |
|---|---|
| Design | Kinetics, https://github.com/UBC-iGEM/internal-wiki-2023-24/commit/2f72daa11b19adc24a234e65ae76012cef7d1d0f |
| Build | |
| Test | |
| Learn |
Project Related Resources
This is where leads and other subteam members can add (or request to notes and resources relating specifically to our project.
Wet Lab
Solid Phase Synthesis
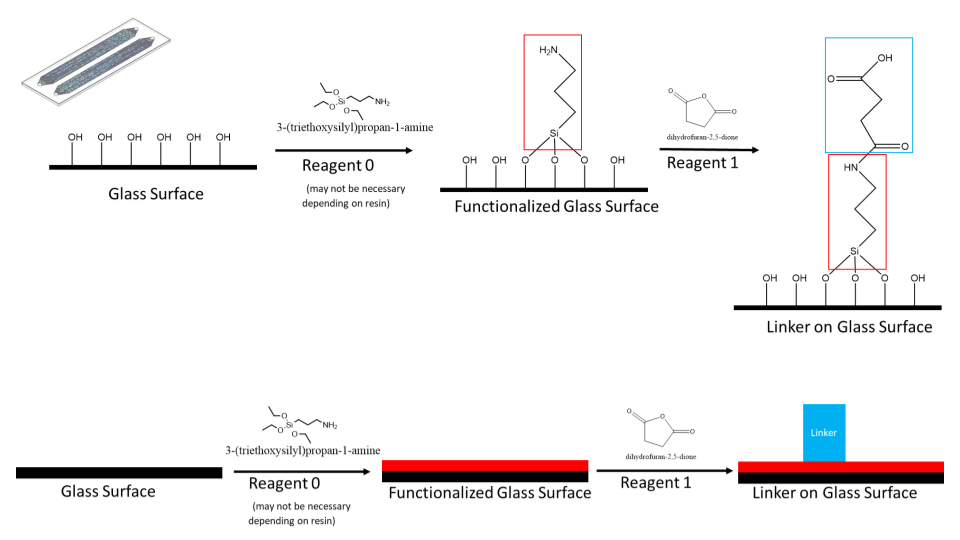
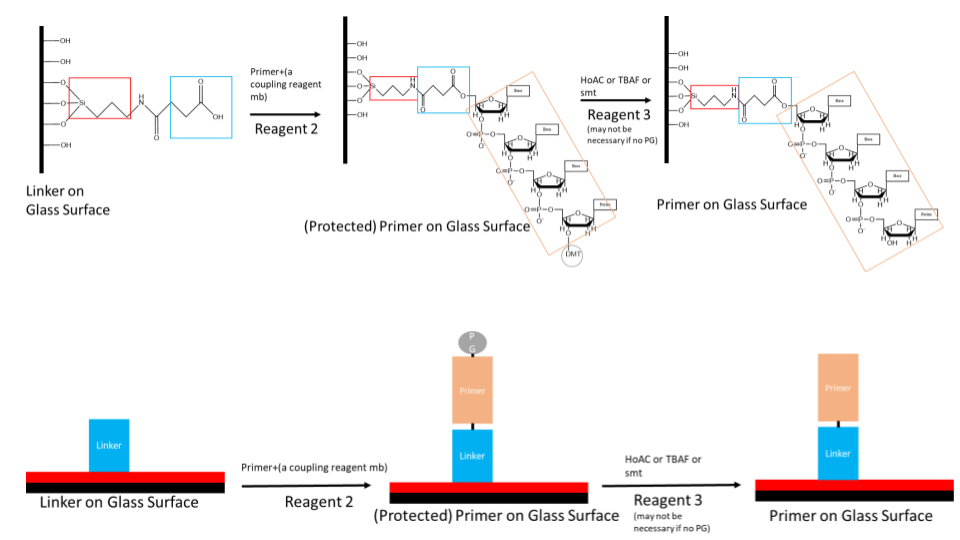
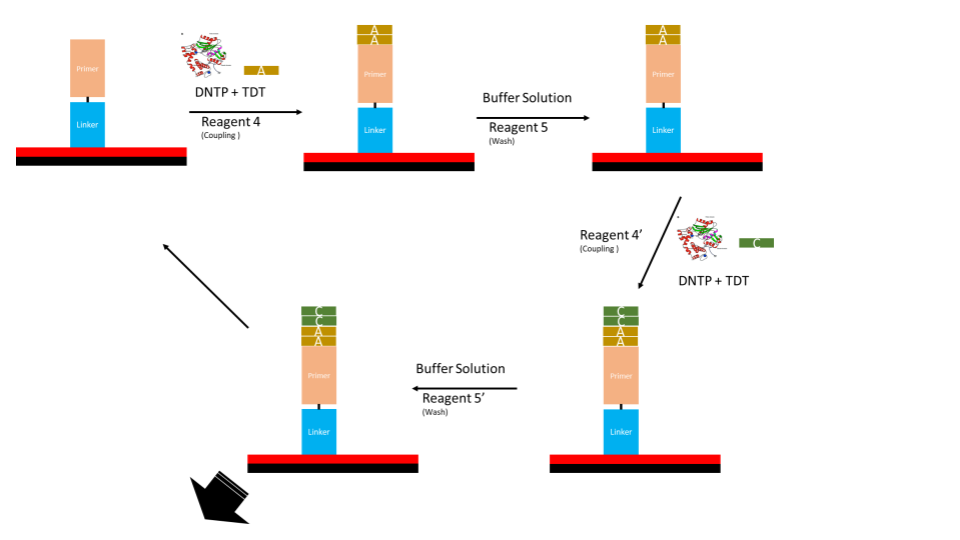
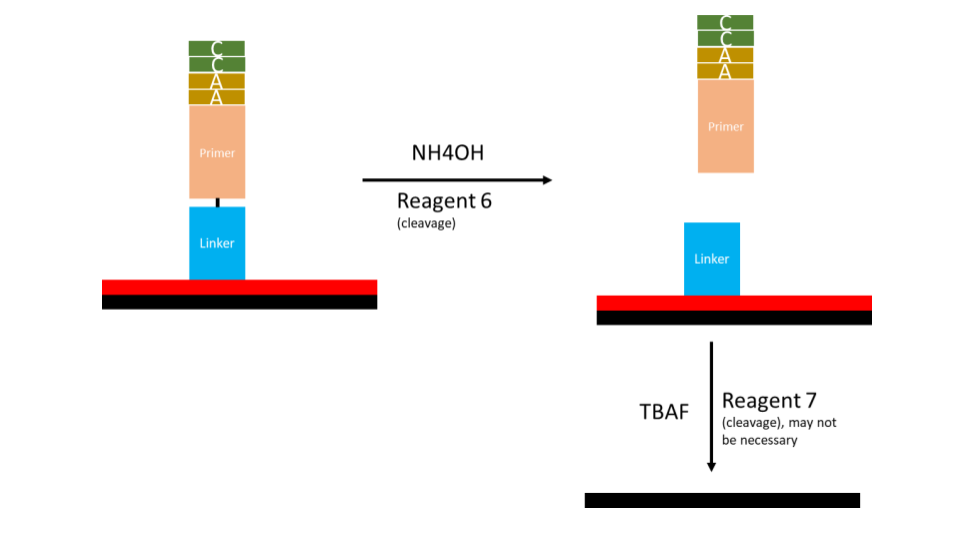
Training Resources
Dry Lab
Slides on Information Theory/Computer Storage
Glacial Storage
Check out these dry lab slides for some info on Glacial Storage!
Information Theory
Why binary?
Error Correction
General Resources
- Introduction to Synthetic Biology
- CSBERG (Canadian Synthetic Biology Education Research Group):
- iGEM engineering webinars (thanks to Kimia for these resources)
- Bioinformatics
- Chemical Engineering Modelling
- Math Modelling
- Enzyme Kinetics
- OrganicChemTutor: What is an Enzyme?
- KhanAcademy: Introduction to Enzyme Kinetics
- KhanAcademy: Steady States and Michaelis-Menten Equations
- KhanAcademy: Enzymatic Inhibition
- KhanAcademy: Cooperativity
- KhanAcademy: Allosteric Regulation and Feedback Loops
- KhanAcademy: Non-Enzymatic Protein Function
- KhanAcademy: Covalent Modifications to Enzymes
- Protein Modelling
Introduction to Synthetic Biology
Thanks to Piyush and Kimia for these resources!
Synthetic biology is a field of biology that combines principles of engineering and science to create new biological systems or modify existing ones for specific purposes. This can include the engineering of new enzymes, the creation of living organisms with specific properties or the development of new genetic circuits. The ultimate goal of synthetic biology is to create new functions or applications that are not found in nature.
To begin, familiarize yourself with general biology. In the suggested videos below, I will be focusing on the central dogma of biology (how do we get from DNA to protein), but I highly recommend you look into additional resources if you need help!
CrashCourse: DNA Structure and Replication
CrashCourse: Transcription and Translation
CrashCourse: What is Biotechnology?
CSBERG (Canadian Synthetic Biology Education Research Group):
iGEM engineering webinars (thanks to Kimia for these resources)
- https://technology.igem.org/engineering/webinars
- https://2021.igem.org/Engineering/Webinars
- Examples of Successful Past iGEM Dry-Lab Projects
Bioinformatics
(thanks to Chae for these resources)
- Analyzing RNA-seq data with DESeq2
- RNA-seq workflow: gene-level exploratory analysis and differential expression
Chemical Engineering Modelling
Math Modelling
Enzyme Kinetics
(Thanks to Piyush for these resources)
In this section, you will be learning about Enzymes, and how they may be modelled mathematically. This is similar to the Le Chatelier’s principle from IB Chemistry 12, so may be a good idea to brush up on that if you are a little rusty!
OrganicChemTutor: What is an Enzyme?
KhanAcademy: Introduction to Enzyme Kinetics
KhanAcademy: Steady States and Michaelis-Menten Equations
KhanAcademy: Enzymatic Inhibition
KhanAcademy: Cooperativity
KhanAcademy: Allosteric Regulation and Feedback Loops
KhanAcademy: Non-Enzymatic Protein Function
KhanAcademy: Covalent Modifications to Enzymes
Protein Modelling
- Solvation free energy of ethanol
- Gromacs Tutorials
- Pyrosetta Tutorials
- Introduction to QM/MM Simulations
- Quantum Mechanics/ Molecular Mechanics (QM/MM)
- Hybrid Quantum-Classical simulations (QM/MM) with CP2K interface
- Machine Learning in QM/MM Molecular Dynamics Simulations of Condensed-Phase Systems
- MiMiC Hybrid Quantum Mechanical/Molecular Mechanical simulations
- Mixed Quantum-Classical simulation techniques
- A user-friendly, Python-based quantum mechanics/Gromacs interface: gmx2qmmm
- Practical: Introduction to quantum mechanics / molecular mechanics (QM/MM) simulations
- Introduction to QM/MM
Welcome
Welcome to the UBC iGEM 2023-2024 Internal Wiki. Read these pages before you start adding to our wiki!
If you have any questions regarding documenting your progress and work, please send a message in the documentation-wiki channel.
What should this internal wiki contain?
This wiki contains everything that lets other members know how other subteams get things running, what other subteams are researching, and the current state of a subteam.
What differentiates this internal wiki from the master todo list? On the master todo list, you can see a task someone in dry lab is working on, while in this internal wiki you can read about the progress, current state, and future plans that member (or subteam) has regarding this task.
On the sidebar you can see all the different categories for each subteam. These folders were put here for a reason; they are a one to one correspondence with the required wiki pages for the iGEM competition. Create files within these folders when adding your content. This internal wiki serves as documentation for every subteam. Any content that is related to any of these topics on the sidebar should be here.
NOTE: We are not writing the final wiki pages here. Later on when we start writing our final content for the iGEM Wiki, use this website as a way to quickly and efficiently absorb and understand material from all our different subteams. The final wiki document is NOT HERE. When you start wiki writing, you will be DIRECTLY committing to the iGEM provisioned Wiki repository on GitLab.
What doesn't this internal wiki contain?
Subteam meeting notes, administrative documents, sponsorship documents, finance. Anything sub-related to our project. Message the documentation channel to verify what goes on the internal wiki if you are unsure. Documentation officially starts when we are finished with pitching and have chosen a project.
Goals
Subteam transparency
Commits to this internal wiki hub are public. This means members can see who is contributing to our documentation and who isn't. The goal is for everyone to be an engaged and active member and contributing to our documentation in a consistent manner is one way to do so. This includes recording down setbacks, progress, blockers, wins and more.
One notable requirement of iGEM is to demonstrate we have used the Engineering Cycle in our project. This means we must demonstrate that we are iterating on the design of our project which is only possible if we record down what went wrong in our processes and how we plan to fix it.
Additionally, iGEM also requires a notebook (from wet lab, but dry lab should also have individual notebooks). Anyone should be able to freely access these notebooks to inquire about what subteams are up to.
We are also using software practices when adding content to our internal wiki. This includes all members making a personal branch to pull updates and push their own, making PRs to merge their content into the main branch, using issues, and more. If you are confused by this, wait until our Git, GitHub and Documentation workshop; if you're still confused send a message in the documentation channel.
Knowledge Transfer
Encourage creativity; the more knowledge sharing we have, the better all our different subteams can collaborate. The more integrated each subteam is the better we will perform at iGEM.
iGEM is a holistic competition. Having a strong dry lab will get you no better than silver if other subteams are struggling. Everyone is expected to have an educated and in depth knowledge of the project from every angle; dry lab, wet lab and HP. This is not possible without documentation from each subteam.
Additionally, teams must integrate each other's work into their own work. This is only possible if knowledge is being shared between all members of the team.
Track Progress: Concrete, small steps (with deliverables) towards our goal
The iGEM project involves many moving parts from all our subteams. In order to ensure everyone is aware of what is going on and to comply with subteam transparency, we must break our project down into a few overarching goals, and then these goals into concrete, small steps. Leads should create goals based on their subteams and larger tasks for these goals, while subteam members should take these larger tasks and break them down into smaller tasks for themselves. This should all be recorded on the internal wiki. Not on Slack channels, not in private DMs or Google Docs. New folders and files should be created to aid this process.
Now you're asking, what does it mean to create a concrete, small step?
Concrete (with a meaningful deliverable)
When defining a next course of action, either for your subteam members or yourself, make sure there is a clear reason for doing this task that contributes to an overarching goal. Each task should also have deliverable that indicates that this step has been successfully completed. For instance, if you are a lead and have given your subteam members a task to understand Golden Gate Assembly, there are two things you should think about. One, why should they learn this procedure? In the context of synthetic biology, you should let your members know that Golden Gate Assembly is an "extremely powerful modular assembly technique in synthetic biology that allows for the efficient and precise assembly of multiple DNA fragments into a single construct." and that we are planning to use this technique in our experiments. Even if you believe the reasoning behind a task is obvious, this is not the case for every member. Be explicit in this reasoning.
Two, how can your subteam members can explicitly demonstrate this understanding in a way that if possible, benefits all members? For instance, ask every member to upload their notes to this internal wiki; then members from other subteams can read what they have written and understand the Golden Gate Assembly as well. This not only allows your members to consolidate their understanding of Golden Gate Assembly, but also to allow knowledge sharing to other subteams.
Small
Tasks should be defined such that they can be completed within a certain time period and contribute to an overarching goal. Time periods that make sense include weekly (to present at our weekly generals) or biweekly; anything more than a month indicates the task may too large or vaguely. If a task you are defining is taking longer than a week or two, then that task can be broken down into smaller tasks. This includes documentation; this documentation should be updated at least once a week from each subteam.
Based on weekly tasks leads give their subteam members, each member should create daily tasks for themselves, that are recorded on this internal wiki. Smaller tasks allow leads to check in on their members to see how they are progressing within the larger weekly task.
Wiki Transfer
Everyone is expected to write on the wiki. Having your progress, results and knowledge here will make wiki writing easier for everyone. The workflow for wiki writing should be as follows:
- You are assigned a page to contribute to.
- You come to this internal wiki to check out related pages.
- If you need more information about a page, you know exactly who to ask by looking at the commit history.
- If your page requires writing about the entire process and not just end results, you can find all of the progress here.
- You start writing the final wiki page in the GitLab repository with this website open.
Questions
Why mdBook?
This documentation format was created by the Rust Language to document their programming language and other Rust projects.
Why aren't we using Notion?
An ideal platform is Notion, however, Canadian schools are not able to get free access to Notion, and our student accounts have limits that cannot accommodate our team of 20 people. Using Notion would cost us $1920.
Why are we using GitHub and Git?
Practice for the final wiki.
Is it okay if I have everything locally?
Brainstorming is okay to keep local, all fleshed out ideas must be on the internal wiki.
Why are we using software/agile practices?
Good way to maintain transparency, and proven to work.
How to use Git and GitHub
- How to add changes to the internal wiki with Codespaces
- How add changes to the internal wiki with Replit
- How to use mdBook locally
How to add changes to the internal wiki with Codespaces
- If you haven't already opened a Codespace, open a new Codespace, otherwise, you can reuse the Codespace you already have open.
- Open up the internal wiki on Codespaces.
- Before you add any changes, run
git pull. This ensures that your local version of the internal wiki is updated with other people's changes. If you don't do this step you make end up with merge conflicts. If runninggit pullresults in merge conflicts (it shouldn't if you're always runninggit pull), let Lucy or a wiki liaison know. - Make a new branch
git branch [name-subteam-description]. Then switch to that branch,git checkout [name-subteam-description]. Someone would run:
git branch lucy-drylab-software
git checkout lucy-drylab-software
- Make your changes in markdown; make sure to preview to ensure your edits are properly formatted.
- When you are ready to publish your updates, you will need to save your files. In git, this is called making a commit. Git provides finer grain control, meaning you can choose which files to save in a project; most of the time you want to save all the files you have modified. Run
git add .. This adds all modified and created files to the staging area. - Once you are satisfied with the files in the staging area, run
git commit -m "[message]". Replace[message]with a meaningful message related to your changes. - Finally, you can run
git push. Make sure you are pushing to the UBC iGEM repo, if asked where to push, push to the repo with the link [https://github.com/UBC-iGEM/internal-wiki-2023-24]; you can push to your own fork, but please enable this setting. - Following the prompts on Codespaces or the GitHub website, make a PR. That's it! Leads and wiki liaisons will check your content and approve and merge your PR.
How add changes to the internal wiki with Replit
How to run the internal wiki on Replit
### How to push changes to internal wiki on ReplitHow to use mdBook locally
If you want to have the full functionality of mdbook, please ask Lucy for instructions on local installation.
How to Document
How to write Markdown
Refer to these documents on how to write markdown if you are using:
Check out the CommonMark quick reference first. Much of this document is borrowed from the rustdoc book[^rust].
Here are some features you must use in our internal wiki:
-
Adding References. See above
-
Adding Tables. To make cooler tables, check out the GitHub Tables extension.
| Header1 | Header2 |
|---|---|
| abc | def |
| Header1 | Header2 |
|---------|---------|
| abc | def |
- Adding task lists.
- [x] Complete task
- [ ] Incomplete task
- Warning blocks
<div class="warning">A big warning!</div>
- Latex, via MathJax
\\[ \mu = \frac{1}{N} \sum_{i=0} x_i \\]
\[ \mu = \frac{1}{N} \sum_{i=0} x_i \]
- Graphs via the DOT Language
Note you can write HTML in Markdown as well! But please refrain from doing so unless necessary.
If you want to see all the Markdown features available to you, here is the spec.
How to add inline citations
- edit src/bibliography.bib, check here for the fields to fill out. For example,
@article{ref_name,
url = {http://www.jstor.org/stable/3263863},
author = {D. J. A. Clines},
journal = {Journal of Biblical Literature},
number = {1},
pages = {22--40},
publisher = {Society of Biblical Literature},
title = {{The Evidence for an Autumnal New Year in Pre-Exilic Israel Reconsidered}},
volume = {93},
year = {1974}
}
- Then call your reference in any mdbook file like this: [@ref_name]
For wiki liaisons
- Why was this role created?
- What is a wiki liaison?
- What are my tasks?
- Working with GitHub
- Who do I ask for help?
- What is my role in developing the competition wiki?
Why was this role created?
This role was created out of frustration from Lucy and Piyush. We noticed a disconnect between subteams, and during wiki development, only a few members could contribute to the wiki because frankly, many members did not know what was happening outside their subteam. This resulted in a wiki that was low effort, unclear, and did not meet the requirements that iGEM judges were looking for.
Wiki liaisons are expected to have a greater understanding of all subteam work, ensure team members (in their own subteam) are contributing to the internal wiki and help Lucy plan for a final competition wiki that aligns with what iGEM is looking for. Wiki liaisons will be a helping hand to the leads, and can offer new perspectives that leads may not notice. Being a wiki liaison will also give you an idea of what a lead's responsibilities are; if you are staying on the team this will help you decide if you want to be a lead. You will help ensure subteam transparency, knowledge transfer and concrete, small steps (with deliverables) towards our goal.
Our wiki liaisons are:
- Wet lab: Diego, Burak
- Dry lab: Riya
- HP: Jessica
What is a wiki liaison?
Generally, a wiki liaison is a subteam member who is not a lead that is helps ensure the work of other subteams are well integrated into with their own subteam's work. This means the wiki liaison helps the wiki lead enforce standards like the internal wiki so that we can meet the three goals listed above. Additionally, wiki liaisons will make sure all information that could contribute to iGEM requirements are being documented, by being proactive in subteam meetings and notifying subteam members to document their findings. They will also have a greater hand in helping design, edit and complete the wiki for the iGEM Jamboree.
What are my tasks?
To have subteam transparency and integration, you will help the wiki lead ensure the entire team is putting their project related content onto the internal wiki, as related to your subteam. This means if you're in wet lab, you make sure all experimental designs and changes are being committed to the internal wiki on a daily/weekly basis, most likely to the wet lab notebook.
Secondly, after each subteam uploads content to the internal wiki, wiki liaisons are responsible for reading other team's content on the internal wiki. Generally, you are trying to form an overall sense of the goals, directions and motivations of the other subteams. This will help you for the third task.
Thirdly, after you have ensured your subteam is contributing to the internal wiki, and have an understanding of what other subteams are doing, you will be responsible for critically analyzing the work of other subteams. For instance, if you are the dry lab liaison, you will have read over the wet lab and HP pages. If the wet lab design pages are hard to understand or missing information, you will create a GitHub issue stating what is missing and why this information is important, and Lucy will alert the appropriate subteam members to add more information. If human practices has described a computational concept incorrectly, or you can enhance the educational material, similarly, make an issue and complete the previously listed steps. You can also make a PR with edits yourself. The point of all this work is to have all our subteams be cohesive and integrated. You are also encouraged to attend other subteam specific meetings, but only if you have the time! For all pages, make sure they are up to date. It's very easy for documentation to fall out of date. If you notice something is no longer correct, alert the appropriate subteam to make changes! Please also read through iGEM documentation before reviewing pages, so you know what you should be looking for.
Finally, when we are writing the wiki for the iGEM Jamboree, you will have a greater hand in helping the leads design the wiki. You may also help assign other subteam members to writing and reading content.
Reviewing wet lab writing
These criteria are nonnegotiable:
- Citations, in the bibliography.bib file
- Related to iGEM requirements
- Sources for information that isn't general knowledge (this is subjective, ask a lead if you need clarification)
- Diagrams for confusing concepts
Reviewing dry lab writing
These criteria are nonnegotiable:
- Citations, in the bibliography.bib file
- Related to iGEM requirements
- For anything code-based: a GitHub repo containing the code in question
- Diagrams for confusing concepts
- Sources for information that isn't general knowledge (this is subjective, ask a lead if you need clarification)
Reviewing human practices writing
- Citations, in the bibliography.bib file
- Related to iGEM requirements
Working with GitHub
You will be reviewing and approving PRs, making issues and sometimes making your own PRs. If you have any questions about this let Lucy now.
Who do I ask for help?
At anytime if you feel this role is overwhelming let the co-directors or Lucy know. We will probably have weekly asynchronous updates on Slack, with meetings if necessary.
If you have any questions that are not of a personal nature, send a message in #wiki-liaison, otherwise DM Lucy.
What is my role in developing the competition wiki?
Based on your subteam, you will help the leads decide how we should present our information, what graphics to make, and more. To be discussed later.